
Abstract
Placental site trophoblastic tumour (PSTT) is a neoplastic proliferation of the implantation intermediate trophoblast. Although clinicopathological studies are not uncommon in case reports or small series, molecular and genetic studies are quite limited. Four archived cases of PSTT were successfully analysed by comparative genomic hybridization (CGH) in this study. Regional chromosomal gains were observed in two cases. One case showed chromosomal gains in the regions of 19p13.2, 21q11–21 and 22q12. The second case demonstrated a single regional chromosomal gain involving 21q21. No chromosomal loss is observed. The remaining two cases showed a balanced CGH profile without detectable chromosomal gain or loss. In summary, although chromosomal alterations detectable by CGH are not common, rare chromosomal gains do occur in PSTT. The recurrent chromosomal gain involving chromosomal 21q observed in two of our cases deserves additional studies to ascertain whether it carries any pathobiological significance.
Similar content being viewed by others


Main
Placental site trophoblastic tumour (PSTT) is a rare gestational trophoblastic disease (GTD), arising from the transformation of implantation intermediate trophoblast following a full-term pregnancy, a molar gestation or a spontaneous abortion.1 In contrast to the highly aggressive, yet treatable gestational choriocarcinoma, PSTT usually behaves in a benign clinical fashion after hysterectomy, but in up to 15% of the cases, the tumour persists and metastasizes. No effective medical treatment for the latter malignant group has been established. The pathogenesis and mechanism of tumour progression of PSTT are largely unknown. Although morphologic and clinical studies are not uncommon in case report or small series, molecular and genetic studies of PSTT are quite limited.2, 3, 4
Comparative genomic hybridization (CGH) is a molecular approach to reach the genome for imbalanced genetic material.5, 6 Labelled genomic test DNA, prepared from tumour sample, is mixed with differently labelled control DNA obtained from normal reference cells. The mixed probes are hybridized to normal metaphase chromosome preparation. Hybridized tester (tumour cells) and control (normal cells) sequences are detected via two different fluorochromes. The ratio of fluorescence intensities for each chromosome can reflect its relative copy number in the tester genome compared with the control genome. Therefore, whole or regional chromosomal gain or loss can be determined. We decided to investigate chromosomal imbalance using CGH in four cases of PSTT.
Materials and methods
Clinical Samples
Four cases were selected from the files of Yale Surgical Pathology with previous established diagnosis of PSTT. The diagnosis was based on well-published histological criteria including immunohistochemical markers. Three patients developed PSTT following a full-term pregnancy and one patient had a complete mole 10 months prior to the development of PSTT. All patients presented with vaginal bleeding and a mildly elevated hCG. Total abdominal hysterectomy and clinical imaging study for extrauterine disease were performed in all four cases. All tumours were routinely fixed in 10% buffered formalin and embedded in paraffin within 24 h after the surgery. The archival age of the specimens ranged from 5 to 8 years. Paraffin blocks containing well-defined tumour area and normal myometrium were selected to extract DNA material for the following CGH study.
Comparative Genomic Hybridization
DNA was isolated from sections of the paraffin blocks from the tumour and the corresponding normal myometrium in each case. Briefly, 10 serial 10 μm sections were cut, with the first one stained with haematoxylin–eosin to verify the presence of tumour tissue, and the remaining nine sections were used for DNA extraction. The areas of interest were outlined and scraped (with caution to avoid unwanted cells) using a blade and collected into a 1.5-ml Eppendorf tube. The paraffin was dissolved by two treatments of 1 ml of xylene at room temperature for 5 min each. The xylene was removed by two washes of 100% ethanol and the deparaffinized tissue samples were air-dried. The DNA was then extracted using the QIAGEN DNA tissue kit (Qiagen, Chatsworth, CA, USA) following the manufacturer's instructions. The concentration of DNA preparation was determined by its absorbance at 260 nm. Using standard nick-translation procedures, test tumour DNA and control DNA were labelled with biotin–11-dUTP and digoxienin–11-dUTP, respectively. The length of the tester and control DNA probes was checked by gel electrophoresis. For optimum hybridization conditions, DNA molecules were kept between 100 and 500 bp in length. The concentration of the DNA probe was measured, and 1:1 mixtures of tester and control labelled probes were prepared for CGH.
For each CGH experiment, a total of 1 μg of mixed probe was used with 25 μg of human Cot-1 DNA (GIBCO/BRL). The mixture was precipitated by ethanol and resuspended into the CGH hybridization buffer containing 50% formamide, 10% dextran sulphate in 2 × SSC:0.3 M NaCl, 30 mM Na citrate, pH 7.0. The probe mixture was denatured at 70°C for 5 min, preannealed at 37°C for 30 min, and then hybridized onto normal male metaphase spreads. Following an incubation in a 37°C moisture chamber for 3 days, the slides were washed and stained with 4,6-diamino-2-phenylindole (DAPI) counter stain.
Image acquisition was performed using an Olympus AX70 Provis microscope equipped with filter sets for an excitation wavelength appropriate to excite one of the three fluorescent molecules. The three images were collected and processed using CytoVision™ computerized imaging system equipped with a COHU monochrome cooled charged-coupled device camera to obtain green/red quantitative fluorescence ratio profile for each chromosome. Profiles from 15 or more metaphase spreads for each specimen were obtained and an average ratio calculated for each chromosome profile. The image representing the DAPI stain was inverted and used for semiautomated karyotyping. Green and red fluorescence intensities were determined from each chromosome from p-telomere to q-telomere by integrating intensities at 1-pixel intervals along the chromosome medial axis. After background correction and normalization of the green–red ratio for each entire metaphase to 1.0, green–red intensity profiles were calculated for all chromosomes. Data from all images were then combined, and an average ratio profile for each chromosome was calculated. A green–red intensity ratio of 0.75 and 1.25 was set as thresholds for under-representation or over-representation of chromosome regions. Profiles at centromeres and close to the telomeric regions were considered unreliable due to in situ suppression in these regions.
Results
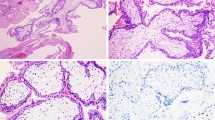
In all four cases of PSTT, the tumour was grossly distinct with nodular expansile mass involving the myometrium (Figure 1a). The average tumour size was 3.5 cm. Histologically, all showed a similar growth pattern with neoplastic cells growing in sheets and cords, permeating the surrounding myometrial smooth muscle (Figure 1b). Tumour DNA material was extracted from the paraffin blocks where the tumour cells constituted over 90% of the cell population. CGH hybridization signals were strong with successful imaging capture in all four cases. Profiles from 15 or more metaphase spreads of each specimen were obtained. Vertical lines on the left side of each chromosome represent loss of genetic material, whereas lines on the right side represent chromosomal gain. The width of the confidence intervals was narrow in all four cases, ensuring a valid CGH hybridization. Two cases (#3 and #4) showed a balanced CGH profile with no chromosomal gain or loss. A regional chromosomal gain at 21q21 was present in case 1 (Figure 1c). The remaining one case (case 2) showed chromosomal gains involving 21q11–21, 19p13.2 and 22q12 (Figure 1d). The magnitude of amplification of these regions was within 1- to 2-fold. No chromosome regional loss was observed in any of the four cases. The presence of hybridization signal to a small number of Y-chromosomes likely represents a nonspecific finding, which has been observed a similar study7 and our previous study using nested genomic PCR failed to demonstrate Y-chromosome-specific sequences in tumour cells of these cases.4 Histologically, the two tumours with abnormal CGH output (cases 1 and 2) showed no significant morphological differences from the other two tumours having a balanced CGH profile (cases 3 and 4). Clinical follow-up information was available in two patients (cases 2 and 4) who were alive without evidence of tumour recurrence.

(a) Gross photograph of a uterus (case 1) involved by a PSTT. A 3.0 cm round tumour mass involves the endometrium and myometrium. (b) Microscopic features of PSTT (case 1) showing sheets of intermediate trophoblasts characterized by large cells with abundant cytoplasm and pleomorphic nuclei, permeating through the adjacent myometrium. (c) CGH profile of PSTT (case 1), showing chromosomal gain involving the chromosomal region at 21q21. (d) CGH profile of PSTT (case 2) showing chromosomal gains involving the chromosomal regions at 19p13.2, 21q11–21, and 22q12.
Discussion
GTD represents a spectrum of trophoblastic proliferations with different growth and malignant potentials,1 including partial hydatidiform mole, complete hydatidiform mole, invasive hydatidiform mole, PSTT, epithelioid trophoblastic tumour and gestational choriocarcinoma. Although our understanding of the pathogenesis of GTDs is limited, an emerging body of data has suggested that abnormal genomic composition and altered epigenic regulation by genomic imprinting appear to play important roles in the development of GTDs.4, 8, 9, 10 It is interesting that, comparable with the proliferative spectrum of GTDs, chromosomal alterations detectable by CGH increase as the lesion becomes more proliferative and aggressive. At the benign end, hydatidiform moles display balanced CGH profiles11 and they carry the least proliferative potential, compatible with a non-neoplastic process. Gestational choriocarcinoma represents the most aggressive GTD and harbours many chromosomal changes detectable by CGH.11 Significant chromosomal gain and loss were reported in nine of 12 cases of choriocarcinoma with recurrent chromosomal deletion at 8p and amplification at 7q. PSTT is a true neoplasm that likely arises from the transformation of intermediate trophoblasts at the implantation site and has an intermediate malignant potential. One recent report analysed two cases of PSTT by CGH and no significant chromosomal gain or loss was found.7 Four archived cases of PSTT were successfully analysed by CGH in this study. Similar to the previous observation, two cases showed a balanced CGH profile. However, rare regional chromosomal gains were observed in two of four cases in our study: one case (case 2) showed chromosomal gains in chromosomal regions of 19p13.2, 21q11–21 and 22q11. The gain involving 21q was a recurrent abnormality, which was observed in another case (case 1), although the regional chromosomal gain was more narrowly mapped to 21q21.
Our study further demonstrated the feasibility of applying CGH on paraffin-archived tissue samples. Degradation of DNA caused by formalin fixation and paraffin embedding, which frequently decreases sensitivity of molecular analysis in routine histological material, appeared not to interfere with CGH analysis in our study. Efficient probe labelling and CGH hybridization were achieved in all four cases. Using paired normal myometrial DNA as a reference probe provides additional specificity of the result as the allotypical genomic variation is completely eliminated. However, CGH permits only a relative gross analysis of chromosomal structures from 3 to 10 Mb.12 Subtle chromosomal deletion or amplification can be missed by CGH study. This may explain a failure to detect chromosomal imbalance in other two cases of PSTT in the current study and a few reported cases,7 as allelic imbalances were not observed infrequently in a recent study.2 Alternatively, the chromosomal regional gains including the recurrent 21q alteration may be unique to the two PSTTs. A regional DNA gain suggests an amplification of potential gene(s) that may provide growth advantage for the tumour.
In summary, CGH is a powerful tool for an initial assessment of genomic abnormalities of human neoplasm and is readily applicable to paraffin-embedded tissue samples. Chromosomal gains or losses detectable by CGH are generally not features of GTDs, except choriocarcinoma. However, rare chromosomal gains do occur in placental site trophoblastic tumour as seen in this study. The recurrent chromosomal gain observed in two of our cases deserves additional studies, such as fluorescent in situ hybridization, to ascertain whether a subtle regional 21q gain can be detected in more cases of PSTT and whether it carries any pathobiological significance.
References
Shih IM, Kurman RJ . The pathology of intermediate trophoblastic tumours and tumour-like lesions. Int J Gynecol Pathol 2001;20:31–47.
Oldt RJ, Kurman RJ, Shih LM . Molecular genetic analysis of placental site trophoblastic tumours and epithelioid trophoblastic tumours confirms their trophoblastic origin. Am J Pathol 2002;161:1033–1037.
Li HW, Tsao SW, Cheung ANY . Current understandings of the molecular genetics of gestational trophoblastic diseases. Placenta 2002;23:20–31.
Hui P, Parkash V, Perkins AS, et al. Pathogenesis of placental site trophoblastic tumour may require the presence of a paternally derived X chromosome. Lab Invest 2000;80:965–972.
Speicher MR, Manoir SD, Schrok E, et al. Molecular cytogenetic analysis of formalin-fixed, paraffin-embedded solid tumours by comparative genomic hybridization after universal DNA-amplification. Hum Mol Gene 1993;2:1907–1914.
Manoir SD, Speicher MR, Joos S, et al. Detection of complete and partial chromosome gains and losses by comparative genomic in situ hybridization. Hum Genet 1993;90:590–610.
Xue WC, Guan XY, Ngan HY, et al. Malignant placental site trophoblastic tumour: a cytogenetic study using comparative genomic hybridization and chromosome in situ hybridization. Cancer 2002;94:2288–2294.
Kajii T, Ohama K . Androgenetic origin of hydatidiform mole. Nature 1977;268:633–634.
Szulman A . Syndromes of hydatidiform moles, partial vs complete. J Reprod Med 1984;29:788–791.
Walsh CM, Miller SJ, Flam F, et al. Paternally derived H19 is differentially expressed malignant and nonmalignant trophoblast. Cancer Res 1995;55:1111–1116.
Ahmed MN, Kim K, Haddad B, et al. Comparative genomic hybridization studies in hydatidiform moles and choriocarcinoma: amplification of 7q21–q31 and loss of 8p12–p21 in choriocarcinoma. Cancer Genet Cytogenet 2000;116:10–15.
Bentx M, Plesch A, Stilgenbauer S, et al. Minimal sizes of deletions detected by comparative genomic hybridization. Genes Chromosomes Cancer 1998;21:172–175.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hui, P., Riba, A., Pejovic, T. et al. Comparative genomic hybridization study of placental site trophoblastic tumour: a report of four cases. Mod Pathol 17, 248–251 (2004). https://doi.org/10.1038/modpathol.3800025
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800025
Keywords
This article is cited by
-
Epithelioid trophoblastic tumor: comparative genomic hybridization and diagnostic DNA genotyping
Modern Pathology (2009)
-
Absence of Y chromosome in human placental site trophoblastic tumor
Modern Pathology (2007)
