
Abstract
Stem cell factor (SCF) and its receptor c-kit take part in the regulation of developmental processes of mast cells, hematopoietic stem cells, and melanocytes, as well as in the growth control of human malignancies. To explore the possible role of the SCF–c-kit system and of mast cells in pancreatic cancer, the concomitant expression and distribution of the two molecules were examined in 17 normal and 26 cancerous human pancreatic tissues and in 6 cultured pancreatic cancer cell lines. Mast cell distribution was also evaluated in the same tissue samples. In addition, the effects of SCF and of the c-kit tyrosine-kinase inhibitor STI571 on the growth of the cancer cell lines and of the normal pancreatic ductal cell line TAKA-1 were assessed. SCF immunoreactivity was absent in acinar, ductal, and islet cells of the normal pancreas and faint in pancreatic cancer tissues and cell lines. In contrast, c-kit was clearly present in some normal and hyperplastic ducts of the normal pancreas, in the cancer cells of 73% of the tumor samples, and in all the cell lines tested. Mast cells, identified by tryptase and chymase immunostaining on consecutive tissue sections, showed immunoreactivity for SCF and c-kit in both normal and cancerous specimens and their number was significantly increased (p = 0.03) in pancreatic cancer compared with the normal pancreas. SCF showed a dose-dependent growth inhibitory effect on TAKA-1 cells (p < 0.001), whereas pancreatic cancer cells were resistant to the SCF-induced growth inhibition. Nonetheless, the growth of TAKA-1 cells and pancreatic cancer cells was inhibited by the c-kit tyrosine kinase inhibitor STI571. In conclusion, the SCF–c-kit system, possibly with the contribution of mast cells, may have a growth-regulating role in the normal pancreas, which is altered during malignant transformation.
Similar content being viewed by others


Introduction
Autocrine and paracrine growth-regulating loops contribute to the aggressiveness of pancreatic carcinoma, one of the most lethal human cancers, the incidence and mortality of which almost coincide (Parker et al, 1997). For example, transforming growth factor β1, β2, and β3 (TGF-β1, TGF-β2, and TGF-β3) isotypes and two of their receptors (TβR-IALK5 and TβRII) are overexpressed in pancreatic cancer (Friess et al, 1993a, 1993b; Lu et al, 1997) and are associated with advanced tumor stage (Lu et al, 1997) and reduced postoperative survival periods (Friess et al, 1993b). Moreover, the concomitant expression of the epidermal growth factor receptor and its ligands by pancreatic cancer cells enhances their aggressiveness and significantly shortens patients’ survival (Korc et al, 1992; Yamanaka et al, 1993). In addition, a number of other alterations of growth-regulating pathways in pancreatic cancer have been described (Ozawa et al, 2001).
The proto-oncogene c-kit encodes a trans-membrane tyrosine-kinase receptor, which is related to the platelet-derived growth-factor/CSF-1 receptor subfamily (Qiu et al, 1988; Yarden et al, 1987). The sequence corresponding to the intracellular domain of c-kit was first identified in the genome of the Hardy-Zuckerman 4 feline sarcoma virus, an acute transforming feline retrovirus (Besmer et al, 1986). The oncogenic potential of c-kit has been demonstrated through its expression in NIH 3T3 mouse fibroblasts (Caruana et al, 1998). After activation of the receptor, enhanced cell proliferation rate and increased capacity of colony formation in soft agar were observed (Caruana et al, 1998).
Stem cell factor (SCF) (Martin et al, 1990), also known as mast cell growth factor (Anderson et al, 1990), steel factor (Witte, 1990), or kit ligand (Huang et al, 1990) has been identified as the ligand of the c-kit receptor on the basis of binding and cross-linking experiments (Huang et al, 1990). The SCF–c-kit pathway is involved in physiologic processes such as gametogenesis, melanogenesis, and hematopoiesis (Geissler et al, 1988). In particular, SCF is the main promoter of mast cell growth and differentiation from bone marrow and peripheral blood progenitors (Valent et al, 1992) and regulates multiple functions of mature mast cells (Bischoff and Dahinden, 1992; Bischoff et al, 1997). Additionally, the SCF–c-kit signaling cascade is thought to contribute to the growth of hematologic and solid malignancies (Coussens et al, 1999; Pietsch et al, 1998), leading to the development of tyrosine-kinase inhibitors as possible treatment options (Mauro et al, 2002).
In this study, the concomitant expression of SCF and c-kit and the distribution of mast cells and their contribution to the production of SCF in pancreatic cancer were assessed. Moreover, the possible role of SCF in the induction of the proliferation of nontransformed ductal pancreatic cells and pancreatic cancer cell lines and the effects of the c-kit tyrosine-kinase inhibitor STI571 on the growth of pancreatic cancer cells were evaluated.
Results
SCF and c-kit Immunostaining in Normal Pancreatic Tissues
In the normal pancreas, SCF immunostaining was confined to the cytoplasm of mononuclear inflammatory cells, which were occasionally present. No immunoreactivity was detected in acinar, ductal, or endocrine cells.
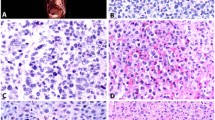
The SCF receptor c-kit was also detected in the cytoplasm and/or on the cell membrane of inflammatory cells. Additionally, moderate to strong c-kit immunoreactivity was present in the cytoplasm of ductal cells of normal and hyperplastic ducts, which were often surrounded by c-kit and, less frequently, SCF-positive inflammatory cells (Fig. 1, A and B). Islet cells occasionally showed weak c-kit immunoreactivity.

A and B, Stem cell factor (SCF) and c-kit immunostaining in normal pancreatic tissues. A duct showing hyperplastic features exhibits strong c-kit-immunoreactivity (A) and it is surrounded by c-kit–positive (A) and SCF-positive (B) inflammatory cells. C, Immunostaining performed with the highly specific anti-tryptase antibody reveals that most of the inflammatory cells described around large and hyperplastic ducts are mast cells (C). Scale bar = 25 μm.
Expression of SCF and c-kit in Pancreatic Cancer
SCF and c-kit immunostaining was evaluated in paraffin-embedded cancer tissue samples. Twelve (80%) of the 15 specimens did not show any SCF immunoreactivity in the cancer cells, whereas in 3 (20%) cases there was a weak to moderate positivity of 5% to 10% of the cancer cells (Fig. 2, A and B). Scattered inflammatory cells were SCF positive.

SCF and c-kit immunostaining in pancreatic cancer tissues. A and B, Focal SCF immunoreactivity of moderately differentiated infiltrating ductal adenocarcinoma. C and D, Diffuse c-kit immunostaining of pancreatic cancer cells. Scale bar = 25 μm (A to C), 50 μm (D).
c-kit expression was widely present in pancreatic cancer specimens, both in cancer cells and, to a lesser extent, in inflammatory cells. Eleven (73%) of 15 cancer samples exhibited weak to strong c-kit immunoreactivity of 5% to 100% of the neoplastic cells (Fig. 2, C and D). As already described in the normal pancreas, c-kit was expressed by ducts showing hyperplastic features, which were occasionally found in the tumor specimens. The four c-kit–negative cases were also SCF negative, but no statistically significant relationship was found between SCF and c-kit expression in pancreatic cancer. In addition, there was no significant relation between SCF or c-kit expression and tumor grading, TNM status, or stage.
Mast Cell Distribution in the Normal Pancreas and Pancreatic Cancer
Tryptase immunostaining was used to detect the presence and the number of mast cells in the normal pancreas and pancreatic cancer tissues. The mean number of tryptase-positive cells in normal tissues, evaluated on paraffin-embedded, formalin-fixed tissue sections and on frozen, Carnoy-fixed tissue sections, was 6.7 ± 3.4/mm2. In cancer samples, the total number of mast cells was significantly higher than in normal tissue, with a mean of 11.1 ± 8.9/mm2 (p = 0.03). Mast cells were located around ducts, blood vessels, and nerves, in the interstitial space between lobules, and, in cancer specimens, in the connective tissue around neoplastic glands (Fig. 3, A and B). However, mast cells did not show a particular clustering around neoplastic cells. To classify the mast cell subtype present in the human pancreas, chymase immunostaining on frozen, Carnoy-fixed, tissue samples was performed (Fig. 3, C and D). The mean number of chymase-positive cells was 1.7 ± 2.1/mm2 in the normal pancreas and 4.7 ± 6.4/mm2 in pancreatic cancer. The difference was not statistically significant.

Tryptase-positive and chymase-positive mast cells in pancreatic cancer tissues. As depicted by tryptase immunostaining, mast cells are located in the connective tissue around neoplastic glands (A and B). Immunostaining performed on consecutive tissue sections shows that in pancreatic cancer, both tryptase-positive and chymase-positive mast cells are present (C and D). Scale bar = 25 μm (B), 50 μm (A, C, and D).
SCF and c-kit Expression by Mast Cells in the Normal Pancreas and Pancreatic Cancer
To identify the SCF-immunoreactive and c-kit-immunoreactive cells observed in the normal and cancerous tissue specimens, consecutive sections stained with tryptase and SCF or c-kit, respectively, were examined. A small percentage (0–13%) of the mast cells present in the normal samples stained positive for SCF. In pancreatic cancer, the number of SCF-expressing mast cells was lower, ranging from 0% to 7% of the total number of mast cells.
c-kit was detected in the cytoplasm and/or on the cell membrane of mast cells in normal pancreas (0–18.5% of total mast cells) and in pancreatic cancer (0–16% of total mast cells). The SCF/c-kit–positive cells described around hyperplastic, c-kit–expressing ducts frequently showed tryptase immunoreactivity (Fig. 1C).
SCF, c-kit, and Tryptase Content in Normal and Malignant Pancreas
SCF, c-kit, and tryptase content was determined by Western blot analysis in four normal and four pancreatic cancer samples. The anti-SCF antibody detected a single band of about 45 kDa in the cancer samples, whereas all the normal pancreatic tissues were negative (Fig. 4). This band corresponds to one of the two described splicing variants of soluble SCF (Pandiella et al, 1992). Analysis of c-kit by Western blotting failed to detect the reported 145-kDa band (Yarden et al, 1987). The size of the c-kit protein varies between human tissues and cell lines (Hassan et al, 1998; Majumder et al, 1988; Sandlow et al, 1997). This may be due to the existence of different splicing isoforms or to differential glycosylation (Lyman and Jacobsen, 1998). Two of the cancer samples displayed a band of approximately 80 kDa, whereas in a third sample a band of about 125 kDa was detected. No bands were seen in any of the normal pancreatic tissues examined (Fig. 4). The 80-kDa band could correspond to the cytoplasmic domain of the c-kit receptor (Besmer et al, 1986; Majumder et al, 1990). The 125-kDa protein has been previously detected by Western blot analysis in human cancer (Tonary et al, 2000).

Western blot analysis of SCF, c-kit, and tryptase in normal pancreas and pancreatic cancer samples, as described in “Materials and Methods.” The highly specific anti-SCF antibody detected a single band of approximately 45 kDa in the cancer samples. The anti-c-kit antibody detected one band of approximately 80 kDa in two of the cancer samples, and a band of about 125 kDa in a third cancer sample. The anti-tryptase antibody detected two bands of approximately 30 and 35 kDa in two pancreatic cancer samples, and a single, intermediate-sized band in one cancer and one normal pancreas sample.
The anti-tryptase antibody detected two bands of about 30 and 35 kDa (as has been previously reported in other tissues; Little and Johnson, 1995) in two pancreatic cancer samples and a single, intermediate-sized band in one cancer and one normal pancreas sample (Fig. 4). In three normal pancreas samples no band was detected.
Expression of SCF and c-kit by TAKA-1 Cells and by Pancreatic Cancer Cell Lines
The next sets of experiments were performed to verify whether the SCF–c-kit system could contribute to the growth stimulation of pancreatic cancer cells. Western blot was used to evaluate SCF and c-kit protein basal level of expression by TAKA-1 cells and by six pancreatic cancer cell lines. TAKA cells did not express SCF, whereas two bands of around 80 kDa and one band of 125 kDa were detected with the c-kit antibody. The two closely spaced 80-kDa bands could correspond to the differently processed cytoplasmic domains of the c-kit receptor. SCF was expressed by Bx-PC-3, MiaPaCa-2, and T3M4 cell lines, the most intense band being present in MiaPaCa-2 cells (Fig. 5). The 80-kDa c-kit proteins were expressed by all the cancer cell lines; Capan-1 showed the highest expression levels (Fig. 5).

Western blot analysis of SCF and c-kit in TAKA-1 cells and in six pancreatic cancer cell lines. TAKA cells did not express SCF, whereas SCF was expressed by Bx-PC-3, MiaPaCa-2, and T3M4 cell lines, the most intense band being present in MiaPaCa-2 cells. The anti-c-kit antibody detected two bands of approximately 80 kDa and one band of 125 kDa in TAKA cells and two bands of approximately 80 kDa in all the cancer cell lines; Capan-1 showed the highest c-kit expression levels.
Localization of c-kit in Pancreatic Cancer Cell Lines and in TAKA-1 Cells
To determine the exact localization of c-kit expression in TAKA-1 and pancreatic cancer cells, flow cytometric analysis was performed next. This analysis revealed that c-kit expression was not detectable in nonpermeabilized cells, whereas a diffuse c-kit positivity was found in permeabilized cells (Fig. 6). This result could indicate the absence of the receptor on the cell membrane of TAKA-1 and pancreatic cancer cells. Alternatively, it could be due to the inability of the antibody to bind its target without entering the cells, because it recognizes the carboxy-terminal, intracellular domain of c-kit.

Flow cytometric detection of c-kit in TAKA-1 and T3M4 pancreatic cancer cell lines. Histograms of cell counts versus fluorescence intensity are shown. Dark histograms indicate the fluorescence profile of TAKA-1 and T3M4 cells after incubation with anti-c-kit antibody, as described in “Materials and Methods;” white histograms represent the negative control. NP = nonpermeabilized; P = permeabilized.
In Vitro Effect of Human Recombinant SCF on the Recovery of Human Intestinal Mast Cells
A previously established SCF-dependent human intestinal mast cell culture assay was used to assess the biologic activity of different human recombinant SCF preparations. SCF purchased from R&D Systems showed a slightly higher potency regarding mast cell recovery compared with SCF derived from Amgen (data not shown). Therefore, SCF from R&D Systems at concentrations between 1 and 100 ng/ml was subsequently used to perform the proliferation assays using TAKA-1 and pancreatic cancer cell lines.
Effect of SCF on the Growth of TAKA-1 Cells and Pancreatic Cancer Cell Lines
SCF showed a dose-dependent growth inhibitory effect on TAKA-1 cells, a threshold effect occurring already at 1 ng/ml. Maximal growth inhibition occurred at 100 ng/ml (p < 0.001). SCF had a modest, although occasionally significant, inhibitory effect on the growth of four of six pancreatic cancer cell lines (Bx-PC-3, Capan-1, COLO 357, Mia-PaCa-2) at different doses (Fig. 7). Maximal inhibition of 20% was observed in Capan-1 cells at dosages of 1 and 2 ng/ml (p = 0.004). To examine whether the effects of SCF on cell growth were mediated through the c-kit receptor, TAKA-1 cells were incubated in the absence or presence of an anti-c-kit antibody that inhibits SCF binding to the receptor. This analysis demonstrated that blockade of the c-kit receptor abolished the growth inhibitory effects of SCF in TAKA-1 cells (Fig. 8).

Effects of SCF on pancreatic cancer cell growth. Cells were incubated in serum-free medium in the absence (control) or presence of the indicated concentrations of SCF (1–100 ng/ml) for 72 hours. Cell growth was measured by the 3-(4,5-methylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay. Percent growth inhibition was determined by comparison with control cell growth. * p < 0.05; ** p < 0.01.

Specificity of the effect of SCF on TAKA-1 cells. TAKA-1 cells were incubated in the absence or presence of SCF (100 ng/ml), anti-c-kit antibody (5 μg/ml), or the combination of both reagents. MTT assay was performed after 72 hours. Percent growth inhibition was determined by comparison with control cell growth. * p < 0.05.
Effect of STI571 on the Growth of TAKA-1 Cells and Pancreatic Cancer Cell Lines
To further clarify the role of the SCF–c-kit system on pancreatic cancer cell growth, in the next set of experiments the c-kit tyrosine kinase inhibitor STI571 was used. STI571 inhibited the growth of TAKA-1 cells and of the three pancreatic cancer cell lines tested in this study in a dose-dependent fashion. Minimal threshold effects of −4% to −9% (p > 0.05) occurred at a concentration of 5 μm STI571. Maximal inhibitory effects of −77% to −90% were observed in these cells at a concentration of 75 μm STI571 (p < 0.001) (Fig. 9).

Effect of STI571 on the growth of TAKA-1 cells and pancreatic cancer cell lines. TAKA-1, ASPC-1, MIA PaCA-2, and T3M4 cells were incubated in the absence (control) or presence of STI571 (in μM) for 72 hours. Cell growth was measured by the MTT assay. Percent growth inhibition was determined by comparison with control cell growth.
Discussion
The present study examines the expression and function of SCF and its tyrosine-kinase receptor c-kit in normal and cancerous pancreatic tissues as well as in pancreatic cell lines.
In the human pancreas, SCF is not expressed by normal exocrine or endocrine cells and only at a very low level by cancer cells. On the other hand, we show that pancreatic mast cells express SCF, which has the potential to inhibit the growth of pancreatic ductal cells. The expression of c-kit by ductal cells shows instead a sort of progression: rare in the normal pancreas, moderate in hyperplastic ducts, present in a high percentage (73%) of cancer specimens, and present in all the cancer cell lines tested in this study. In both normal and cancer specimens, immunohistochemistry performed on consecutive sections showed that the mononuclear inflammatory cells expressing SCF and c-kit are mostly tryptase/chymase double-positive mast cells (MCTC), also named connective tissue-type mast cells (Irani et al, 1986). Although it is well known that human mast cells express c-kit and SCF (Tsuura et al, 1994; Zhang et al, 1998), this is the first report that characterizes the presence of mast cells in pancreatic cancer tissue in a significantly higher number than in the normal pancreas. Based on our findings, it is tempting to speculate that the recruitment of mast cells into the neoplastic tissue may be induced by the cancer cells expressing SCF. Indeed, there is now accumulating evidence that mast cells may regulate tumor growth and invasion by different means. For example, mast cells produce growth stimulating and proangiogenic factors that enhance the growth of lung and skin cancers (Coussens et al, 1999; Imada et al, 2000). In pulmonary adenocarcinoma, the mast cell number correlates with the presence of lymph node metastases and advanced tumor stage (Takanami et al, 2000). In breast cancer, the concentration of mast cells at the tumor edge suggested a possible role for mast cell-derived proteases in the degradation of the extracellular matrix and tumor invasion (Kankkunen et al, 1997). However, a relation between mast cell number and stage of the disease was not found in this series of pancreatic carcinomas.
The analysis of the expression of c-kit and of its ligand SCF in human neoplasms yielded some conflicting results. Simultaneous expression of c-kit and SCF has been reported in colon, gastric, and lung cancer cell lines, in which a growth/migration-stimulating autocrine loop has been demonstrated (Bellone et al, 1997; DiPaola et al, 1997; Hassan et al, 1998). On the other hand, in melanomas and in breast and thyroid carcinomas a loss of expression of these molecules, in comparison with normal melanocytes and mammary or thyroid epithelium, has been described (Moretti et al, 1999; Natali et al, 1992, 1995; Welker et al, 2000). The introduction of a c-kit expression vector in melanoma and breast cancer cell lines caused growth inhibition (Huang et al, 1996; Nishida et al, 1996). Additionally, the exposure of the c-kit–transfected melanoma cell line to SCF triggered apoptosis of these cells but not of the parental, c-kit–negative cell line (Huang et al, 1996).
Although the mechanism of the SCF growth inhibitory effect on normal pancreatic ductal cells described in this study has not been elucidated, it is possible that the SCF–c-kit growth regulating pathway acts as a negative control element in non-neoplastic pancreatic ductal epithelium, whereas this function is lost in the transformed ducts. The presence of SCF-producing mast cells surrounding hyperplastic, c-kit–expressing pancreatic ducts favors the hypothesis of a growth-regulating rather than oncogenic potential of SCF in human pancreatic tissue. Moreover, our finding of a clear, dose-dependent, and specific growth inhibitory effect of SCF on cultured TAKA-1 cells gives further support to this concept. TAKA-1 cells are obtained from normal pancreatic ductal cells of the hamster and bear chromosome 3 alterations that induce immortalization (Takahashi et al, 1995a) but do not involve genes related to pancreatic tumorigenesis (Takahashi et al, 1995b). Therefore, even though they may not exactly reproduce the characteristics of normal human pancreatic ductal cells, they seem to be an appropriate non-neoplastic control in experiments performed on pancreatic cancer cell lines (Kleeff et al, 1999).
An explanation for the loss of SCF-induced growth inhibition in pancreatic cancer cell lines might be the presence of a smaller c-kit protein compared with that found in TAKA-1 cells (Fig. 5). The 80-kDa c-kit proteins detected in pancreatic cancer cell lines could correspond to truncated, nonfunctional proteins, as has been previously described in gastric and colon cancer cell lines (Hassan et al, 1998; Toyota et al, 1994).
The evaluation of the expression of protein-kinase receptors and their ligands in human cancers is of particular interest because selective protein-kinase inhibitors have been developed (Druker and Lydon, 2000). One of these molecules, the tyrosine kinase inhibitor STI571, has been shown to inhibit BCR/ABL, TEL/ABL, v-Abl, ARG, platelet-derived growth factor β receptor, and c-kit (Okuda et al, 2001). Preclinical and clinical studies have shown the ability of STI571 to exert significant cytotoxic effects in chronic myeloid leukemia, which expresses the BCR/ABL tyrosine kinase (Mauro and Druker, 2001), and in small-cell lung cancers and gastrointestinal stromal tumors, which express c-kit in a high percentage of cases (Joensuu et al, 2001; Krystal et al, 2000). In this study, the administration of STI571 to three pancreatic cancer cell lines resulted in a significant growth inhibition, although only at relatively high concentrations. While the exact mechanisms of this effect are not known, there are two alternative explanations. First, STI571 might act through a c-kit–independent signaling cascade by blocking the phosphorylation of other tyrosine kinase receptors (eg, platelet-derived growth factor β receptor), which have the capacity to transmit mitogenic signals in pancreatic cancer (Ebert et al, 1995). Alternatively, blockage of c-kit receptor phosphorylation might directly inhibit pancreatic cancer cell growth through currently unknown mechanisms, which are independent of a functional SCF–c-kit signaling pathway. Irrespective of the exact mechanisms of the function of STI571 in pancreatic cancer, the observed growth inhibitory effects of this tyrosine kinase inhibitor—although at concentrations severalfold higher than currently achieved in clinical settings—suggest that this compound warrants further research regarding its potential use in the treatment of pancreatic cancer in the future.
CONCLUSION
In summary, we show a significant increase of tryptase-producing mast cells in human pancreatic cancers and coexpression of SCF and c-kit in a limited percentage of cases. Pancreatic cancer cells might have the capacity to recruit mast cells to the tumor mass via SCF production; thereafter, mast cells might potentiate tumor invasion and angiogenesis through their effects on the extracellular matrix. In addition, mast cells—through the production of SCF—could directly participate in the regulation of the growth of pancreatic ductal cells, which become resistant to the inhibitory effects of SCF after malignant transformation. In conclusion, the interaction between mast cells and the SCF–c-kit system might play an important role in the pathophysiology of pancreatic cancer.
Materials and Methods
The following materials were purchased: FCS, RPMI and DMEM, trypsin-EDTA solution, and penicillin-streptomycin solution from Biochrom KG (Berlin, Germany); human recombinant SCF from R&D Systems (Abingdon, United Kingdom) and from Amgen GmbH (Munich, Germany); anti-SCF antibody from Santa Cruz Biotechnology (Santa Cruz, California); anti-c-kit polyclonal antibodies from Santa Cruz Biotechnology and from NeoMarkers (Fremont, California); anti-tryptase and anti-chymase monoclonal antibodies from Chemicon International (Temecula, California); biotinylated secondary antibodies and streptavidin/peroxidase complex from Kierkegaard and Perry Laboratories (Gaithersburg, Maryland); FITC-conjugated anti-rabbit antibody from Zymed (San Francisco, California); OCT compound from Miles Inc. (Elkhart, Indiana); horseradish peroxidase-conjugated secondary antibodies and ECL Western blotting detection reagents from Amersham Life Science (Amersham, United Kingdom); and Protease Inhibitor Cocktail Tablets from Roche (Basel, Switzerland). All other reagents were from Sigma Chemical Company (St. Louis, Missouri). ASPC-1, Bx-PC-3, Capan-1, MIA PaCA-2, and human pancreatic cell lines were obtained from American Type Culture Collection (Rockville, Maryland). STI571 was kindly provided by Novartis Pharma AG (Basel, Switzerland). COLO 357 and T3M4 human pancreatic cell lines were a gift from Dr. R. S. Metzgar (Durham, North Carolina). TAKA-1 immortalized Syrian golden hamster pancreatic ductal cells were a gift from Prof. P. Pour (University of Nebraska, Omaha, Nebraska).
Tissue Samples
Normal human pancreatic tissue samples (n = 17; 11 men, 6 women; median age, 46 years; range, 18–77 years) and pancreatic cancer tissues (n = 26; 10 men, 16 women; median age, 69 years; range, 35–83 years) were obtained through an organ donor program and from patients undergoing surgery for primary pancreatic cancer. In 22 patients (84%) a pancreatico-duodenectomy was performed; 3 patients (12%) underwent a pancreatic left resection, and in 1 patient (4%) a subtotal pancreatectomy was performed.
Tissue samples for histologic and immunohistochemical analysis were collected freshly in the operating room, fixed immediately in 10% formalin for 12 to 24 hours and embedded in paraffin or embedded in OCT, and snap-frozen in liquid nitrogen. In addition, tissue samples were frozen in liquid nitrogen immediately upon surgical removal and kept at −80° C until use for protein extraction. Histologic examination revealed that the normal pancreatic specimens, which were obtained through an organ donor program, were all non-neoplastic, noninflammatory pancreatic tissues that contained occasionally hyperplastic, but typical, ductal structures. Staging and grading of the cancer specimens was assessed according to the TNM classification of the International Union Against Cancer. All the tumor samples were ductal adenocarcinomas; 7 (27%) were well differentiated (G1), 14 (54%) were moderately differentiated (G2), and 5 (19%) were poorly differentiated (G3). Four tumors (15%) were in Stage I, 5 (19%) were in Stage II, 15 (58%) were in Stage III, and 2 (8%) were in Stage IV. All studies were approved by the Human Subjects Committee of the University of Bern, Switzerland.
Immunohistochemistry
Consecutive paraffin-embedded tissue sections (12 normal pancreas and 15 pancreatic cancer samples; 2- to 4-μm thick) were subjected to immunostaining using the streptavidin-peroxidase technique. After deparaffinization and hydration, the slides were washed in Tris-buffered saline (TBS) (10 mm Tris HCl, 0.85% NaCl, pH 7.4) containing 0.1% (w/v) BSA. Endogenous peroxidase activity was quenched by incubating the slides in methanol and methanol/0.6% hydrogen peroxide, followed by washings in methanol and in TBS-0.1% BSA.
The following primary antibodies directed against human proteins were used: goat anti-SCF (1:100 dilution), rabbit anti-c-kit (1:200 dilution), and, to detect the presence of mast cells, mouse anti-tryptase (1:1000 dilution) and anti-chymase (1:4000 dilution). For SCF immunostaining, the sections were pretreated with pronase (1 mg/ml in TBS, pH 7.5) for 6 minutes at 37° C on a warming plate, for antigen retrieval. Cryostat sections obtained from OCT-embedded tissues (5 normal pancreas and 11 pancreatic cancer samples) were fixed for 5 minutes at 4° C in Carnoy’s solution and used for chymase detection, because formalin fixation destroys chymase immunoreactivity (Konttinen et al, 2000). Consecutive sections of the same frozen samples were stained for tryptase, to compare the number of tryptase-positive and chymase-positive cells in the same tissue samples.
After incubation with primary antibodies, slides were washed in TBS buffer and then appropriate biotinylated secondary antibodies were added at room temperature for 45 minutes. After washing in TBS buffer and incubation with the streptavidin/peroxidase complex at room temperature for 30 minutes, the slides were incubated with diaminobenzidine tetrahydrochloride solution in Tris-HCl buffer (0.05 g/100 ml, pH 7.6) containing 0.03% hydrogen peroxide as the substrate and counterstained with Mayer’s hematoxylin.
To ensure antibody specificity, consecutive sections were incubated in the absence of primary antibody. In these cases no specific immunostaining was detected.
Evaluation of Immunohistochemical Findings
SCF and c-kit expression of the cancer samples was quantified by counting at least 1000 cells in each tumor sample. Immunohistochemical findings were scored positive when at least 5% of the neoplastic cells were immunoreactive (Arber et al, 1998).
A picture of each section was taken using a high-resolution color video camera on a light microscope. The area of the whole section was determined using a computer-assisted image analysis system (Image-Pro-Plus for Windows, version 4.0; Media Cybernetics, Silver Spring, Maryland). The number of tryptase-positive cells and, in the frozen sections, of tryptase-positive and chymase-positive cells was counted at a × 200 magnification and expressed as number of cells per square millimeter. The number of SCF-positive and c-kit–positive mast cells was expressed as a percentage of total mast cells present in the paraffin-embedded tissue specimens, by comparing two consecutive sections stained with anti-tryptase and anti-SCF or anti-tryptase and anti-c-kit, respectively.
Western Blot Analysis
Proteins were extracted from approximately 200 mg of frozen normal and cancer tissues. Cells were lysed in a buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA, and 1% SDS. Protein concentrations were measured with the micro BCA protein assay (Pierce, Rockford, Illinois). Fifty micrograms of protein from each sample of normal pancreas or cancer tissues and 30 μg of protein from cell lysates were separated on SDS-polyacrylamide gels and electroblotted onto nitrocellulose membranes. Membranes were then incubated in blocking solution (5% nonfat milk in 20 mm Tris HCl, 150 mm NaCl, 0.1% Tween-20) (TBS-T), followed by incubation with anti-SCF, anti-c-kit, anti-tryptase, or anti-chymase antibodies. The membranes were then washed in TBS-T and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Antibody detection was performed with an enhanced chemiluminescence reaction. To ensure equivalent loading and transfer, membranes were routinely stained with Amido Black.
Cytofluorimetric Detection of c-kit
TAKA-1, ASPC-1, MIA PaCA-2, and T3M4 cell lines were tested for the presence of surface and/or cytoplasmic c-kit. Cells (5 × 105) were harvested, washed twice in PBS containing 0.1% BSA, and then incubated with rabbit anti-c-kit antibody (1:10) for 30 minutes at room temperature. After washing as above, cells were incubated with FITC-conjugated goat anti-rabbit antibody (1:20) for 30 minutes at 4C°, then washed again and analyzed with a FACSort flow cytometer (Becton Dickinson, Le Pont de Claix, France). To detect cytoplasmic c-kit, cells were permeabilized by washing in PBS containing 0.1% BSA and 0.2% Triton X before incubation with the primary antibody. Omission of the primary antibody was used as a negative control.
Cell Culture and Proliferation Assay
Human pancreatic cancer cell lines were routinely grown in DMEM (Bx-PC-3, COLO 357, and MIA PaCA-2) or RPMI (ASPC-1, Capan-1, T3M4) supplemented with 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin (complete medium). To choose an appropriate range of dose for the human recombinant SCF to be used for the proliferation assays, SCF from different sources (R&D and Amgen) were tested on a well-established cell culture model of isolated human intestinal mast cells (see below).
To perform growth assays, cells were seeded overnight at a density of 2000 cells/well in 96-well plates and subsequently incubated in serum-free medium (DMEM/RPMI containing 0.1% BSA, 5 μg/ml apotransferrin, 5 ng/ml sodium selenite, and antibiotics) in the absence or presence of SCF at dosages of 1, 2, 10, 50, and 100 ng/ml. After 72 hours, 3-(4,5-methylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was added (50 μg/well) for 4 hours. Cellular MTT was solubilized with acidic isopropanol, and the optical density was measured at 570 nm using a plate reader (Dynatech MR 7000; Dynatech Labs, Chantilly, VA).
TAKA-1 cells were grown in RPMI supplemented with 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were seeded overnight at a density of 5000 cells/well in 96-well plates, and the proliferation assay was performed as described above. To assess the specificity of the cell response to SCF, a growth assay was performed in TAKA-1 cells in the presence of an anti-c-kit antibody that inhibits the binding of SCF to its receptor. Briefly, cells were seeded overnight in 96-well plates and subsequently incubated in the presence of SCF (100 ng/ml), the anti-c-kit antibody (5 μg/ml), and the combination of both reagents, respectively. MTT was performed after 72 hours. To assess the effect of STI571 on cell growth, TAKA-1 cells and pancreatic cancer cell lines were incubated in the absence (control) or presence of the indicated concentrations of STI571. Cell growth was determined after 72 hours by the MTT colorimetric assay. All experiments were performed in triplicate.
Mast Cell Culture
Human mast cells were isolated from intestinal surgery specimens and purified to homogeneity as described in detail elsewhere (Bischoff et al, 1997, 1999). The purification procedure of mast cells consists of a positive selection of c-kit–expressing cells using magnetic cell separation (MACS system; Miltenyi Biotec, Bergisch Gladbach, Germany) and the mAb YB5.B8 (PharMingen, Hamburg, Germany) directed against human c-kit and subsequent culture of the selected cells for 14 days in standard medium (RPMI 1640) supplemented with human recombinant SCF from different sources (Amgen and R&D). Without SCF all mast cells died within a few days. In the presence of SCF, mast cell purity reached 100% after 14 days of culture. Mast cell recovery was calculated by counting mast cell numbers at culture start (MCstart) and after culture (MCend) using the formula 100 × MCend/MCstart.
Statistical Analysis
Results were expressed as mean ± sd. For statistical analysis, the Student’s t test was used. Significance was defined as p < 0.05.
References
Anderson DM, Lyman SD, Baird A, Wignall JM, Eisenman J, Rauch C, March CJ, Boswell HS, Gimpel SD, and Cosman D (1990). Molecular cloning of mast cell growth factor, a hematopoietin that is active in both membrane bound and soluble forms. Cell 63: 235–243.
Arber DA, Tamayo R, and Weiss LM (1998). Paraffin section detection of the c-kit gene product (CD117) in human tissues: Value in the diagnosis of mast cell disorders. Hum Pathol 29: 498–504.
Bellone G, Silvestri S, Artusio E, Tibaudi D, Turletti A, Geuna M, Giachino C, Valente G, Emanuelli G, and Rodeck U (1997). Growth stimulation of colorectal carcinoma cells via the c-kit receptor is inhibited by TGF-beta 1. J Cell Physiol 172: 1–11.
Besmer P, Murphy JE, George PC, Qiu FH, Bergold PJ, Lederman L, Snyder HW, Brodeur D, Zuckerman EE, and Hardy WD (1986). A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature 320: 415–421.
Bischoff SC and Dahinden CA (1992). c-kit ligand: A unique potentiator of mediator release by human lung mast cells. J Exp Med 175: 237–244.
Bischoff SC, Schwengberg S, Raab R, and Manns MP (1997). Functional properties of human intestinal mast cells cultured in a new culture system: Enhancement of IgE receptor-dependent mediator release and response to stem cell factor. J Immunol 159: 5560–5567.
Bischoff SC, Sellge G, Lorentz A, Sebald W, Raab R, and Manns MP (1999). IL-4 enhances proliferation and mediator release in mature human mast cells. Proc Natl Acad Sci USA 96: 8080–8085.
Caruana G, Cambareri AC, Gonda TJ, and Ashman LK (1998). Transformation of NIH3T3 fibroblasts by the c-Kit receptor tyrosine kinase: Effect of receptor density and ligand-requirement. Oncogene 16: 179–190.
Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, and Hanahan D (1999). Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev 13: 1382–1397.
DiPaola RS, Kuczynski WI, Onodera K, Ratajczak MZ, Hijiya N, Moore J, and Gewirtz AM (1997). Evidence for a functional kit receptor in melanoma, breast, and lung carcinoma cells. Cancer Gene Ther 4: 176–182.
Druker BJ and Lydon NB (2000). Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest 105: 3–7.
Ebert M, Yokoyama M, Friess H, Kobrin MS, Buchler MW, and Korc M (1995). Induction of platelet-derived growth factor A and B chains and over-expression of their receptors in human pancreatic cancer. Int J Cancer 62: 529–535.
Friess H, Yamanaka Y, Buchler M, Berger HG, Kobrin MS, Baldwin RL, and Korc M (1993a). Enhanced expression of the type II transforming growth factor beta receptor in human pancreatic cancer cells without alteration of type III receptor expression. Cancer Res 53: 2704–2707.
Friess H, Yamanaka Y, Buchler M, Ebert M, Beger HG, Gold LI, and Korc M (1993b). Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 105: 1846–1856.
Geissler EN, Ryan MA, and Housman DE (1988). The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell 55: 185–192.
Hassan S, Kinoshita Y, Kawanami C, Kishi K, Matsushima Y, Ohashi A, Funasaka Y, Okada A, Maekawa T, He-Yao W, and Chiba T (1998). Expression of protooncogene c-kit and its ligand stem cell factor (SCF) in gastric carcinoma cell lines. Dig Dis Sci 43: 8–14.
Huang S, Luca M, Gutman M, McConkey DJ, Langley KE, Lyman SD, and Bar-Eli M (1996). Enforced c-KIT expression renders highly metastatic human melanoma cells susceptible to stem cell factor-induced apoptosis and inhibits their tumorigenic and metastatic potential. Oncogene 13: 2339–2347.
Huang E, Nocka K, Beier DR, Chu TY, Buck J, Lahm HW, Wellner D, Leder P, and Besmer P (1990). The hematopoietic growth factor KL is encoded by the Sl locus and is the ligand of the c-kit receptor, the gene product of the W locus. Cell 63: 225–233.
Imada A, Shijubo N, Kojima H, and Abe S (2000). Mast cells correlate with angiogenesis and poor outcome in stage I lung adenocarcinoma. Eur Respir J 15: 1087–1093.
Irani AA, Schechter NM, Craig SS, DeBlois G, and Schwartz LB (1986). Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci USA 83: 4464–4468.
Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, Silberman S, Capdeville R, Dimitrijevic S, Druker B, and Demetri GD (2001). Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 344: 1052–1056.
Kankkunen JP, Harvima IT, and Naukkarinen A (1997). Quantitative analysis of tryptase and chymase containing mast cells in benign and malignant breast lesions. Int J Cancer 72: 385–388.
Kleeff J, Maruyama H, Ishiwata T, Sawhney H, Friess H, Buchler MW, and Korc M (1999). Bone morphogenetic protein 2 exerts diverse effects on cell growth in vitro and is expressed in human pancreatic cancer in vivo. Gastroenterology 116: 1202–1216.
Konttinen YT, Hietanen J, Virtanen I, Ma J, Sorsa T, Xu JW, Williams NP, Manthorpe R, and Janin A (2000). Mast cell derangement in salivary glands in patients with Sjögren’s syndrome. Rheumatol Int 19: 141–147.
Korc M, Chandrasekar B, Yamanaka Y, Friess H, Buchier M, and Beger HG (1992). Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J Clin Invest 90: 1352–1360.
Krystal GW, Honsawek S, Litz J, and Buchdunger E (2000). The selective tyrosine kinase inhibitor STI571 inhibits small cell lung cancer growth. Clin Cancer Res 6: 3319–3326.
Little SS and Johnson DA (1995). Human mast cell tryptase isoforms: Separation and examination of substrate-specificity differences. Biochem J 307: 341–346.
Lu Z, Friess H, Graber HU, Guo X, Schilling M, Zimmermann A, Korc M, and Buchler MW (1997). Presence of two signaling TGF-beta receptors in human pancreatic cancer correlates with advanced tumor stage. Dig Dis Sci 42: 2054–2063.
Lyman SD and Jacobsen SE (1998). c-kit ligand and Flt3 ligand: Stem/progenitor cell factors with overlapping yet distinct activities. Blood 91: 1101–1134.
Majumder S, Brown K, Qiu FH, and Besmer P (1988). c-kit protein, a transmembrane kinase: Identification in tissues and characterization. Mol Cell Biol 8: 4896–4903.
Majumder S, Ray P, and Besmer P (1990). Tyrosine protein kinase activity of the HZ4-feline sarcoma virus P80gag-kit-transforming protein. Oncogene Res 5: 329–335.
Martin FH, Suggs SV, Langley KE, Lu HS, Ting J, Okino KH, Morris CF, McNiece IK, Jacobsen FW, and Mendiaz EA (1990). Primary structure and functional expression of rat and human stem cell factor DNAs. Cell 63: 203–211.
Mauro MJ and Druker BJ (2001). Sti571: A gene product-targeted therapy for leukemia. Curr Oncol Rep 3: 223–227.
Mauro MJ, O’Dwyer M, Heinrich MC, and Druker BJ (2002). STI571: A paradigm of new agents for cancer therapeutics. J Clin Oncol 20: 325–334.
Moretti S, Pinzi C, Spallanzani A, Berti E, Chiarugi A, Mazzoli S, Fabiani M, Vallecchi C, and Herlyn M (1999). Immunohistochemical evidence of cytokine networks during progression of human melanocytic lesions. Int J Cancer 84: 160–168.
Natali PG, Berlingieri MT, Nicotra MR, Fusco A, Santoro E, Bigotti A, and Vecchio G (1995). Transformation of thyroid epithelium is associated with loss of c-kit receptor. Cancer Res 55: 1787–1791.
Natali PG, Nicotra MR, Sures I, Mottolese M, Botti C, and Ullrich A (1992). Breast cancer is associated with loss of the c-kit oncogene product. Int J Cancer 52: 713–717.
Nishida K, Tsukamoto T, Uchida K, Takahashi T, and Ueda R (1996). Introduction of the c-kit gene leads to growth suppression of a breast cancer cell line, MCF-7. Anticancer Res 16: 3397–3402.
Okuda K, Weisberg E, Gilliland DG, and Griffin JD (2001). ARG tyrosine kinase activity is inhibited by STI571. Blood 97: 2440–2448.
Ozawa F, Friess H, Tempia-Caliera A, Kleeff J, and Buchler MW (2001). Growth factors and their receptors in pancreatic cancer. Teratog Carcinog Mutagen 21: 27–44.
Pandiella A, Bosenberg MW, Huang EJ, Besmer P, and Massague J (1992). Cleavage of membrane-anchored growth factors involves distinct protease activities regulated through common mechanisms. J Biol Chem 267: 24028–24033.
Parker SL, Tong T, Bolden S, and Wingo PA (1997). Cancer statistics, 1997. CA Cancer J Clin 47: 5–27.
Pietsch T, Nicotra MR, Fraioli R, Wolf HK, Mottolese M, and Natali PG (1998). Expression of the c-Kit receptor and its ligand SCF in non-small-cell lung carcinomas. Int J Cancer 75: 171–175.
Qiu FH, Ray P, Brown K, Barker PE, Jhanwar S, Ruddle FH, and Besmer P (1988). Primary structure of c-kit: Relationship with the CSF-1/PDGF receptor kinase family–oncogenic activation of v-kit involves deletion of extracellular domain and C terminus. EMBO J 7: 1003–1011.
Sandlow JI, Feng HL, and Sandra A (1997). Localization and expression of the c-kit receptor protein in human and rodent testis and sperm. Urology 49: 494–500.
Takahashi T, Moyer MP, Cano M, Wang QJ, Adrian TE, Mountjoy CP, Sanger W, Sugiura H, Katoh H, and Pour PM (1995a). Establishment and characterization of a new, spontaneously immortalized, pancreatic ductal cell line from the Syrian golden hamster. Cell Tissue Res 282: 163–174.
Takahashi T, Moyer MP, Cano M, Wang QJ, Mountjoy CP, Sanger W, Adrian TE, Sugiura H, Katoh H, and Pour PM (1995b). Differences in molecular biological, biological and growth characteristics between the immortal and malignant hamster pancreatic cells. Carcinogenesis 16: 931–939.
Takanami I, Takeuchi K, and Naruke M (2000). Mast cell density is associated with angiogenesis and poor prognosis in pulmonary adenocarcinoma. Cancer 88: 2686–2692.
Tonary AM, Macdonald EA, Faught W, Senterman MK, and Vanderhyden BC (2000). Lack of expression of c-KIT in ovarian cancers is associated with poor prognosis. Int J Cancer 89: 242–250.
Toyota M, Hinoda Y, Itoh F, Takaoka A, Imai K, and Yachi A (1994). Complementary DNA cloning and characterization of truncated form of c-kit in human colon carcinoma cells. Cancer Res 54: 272–275.
Tsuura Y, Hiraki H, Watanabe K, Igarashi S, Shimamura K, Fukuda T, Suzuki T, and Seito T (1994). Preferential localization of c-kit product in tissue mast cells, basal cells of skin, epithelial cells of breast, small cell lung carcinoma and seminoma/dysgerminoma in human: Immunohistochemical study on formalin-fixed, paraffin-embedded tissues. Virchows Arch 424: 135–141.
Valent P, Spanblochl E, Sperr WR, Sillaber C, Zsebo KM, Agis H, Strobl H, Geissler K, Bettelheim P, and Lechner K (1992). Induction of differentiation of human mast cells from bone marrow and peripheral blood mononuclear cells by recombinant human stem cell factor/kit-ligand in long-term culture. Blood 80: 2237–2245.
Welker P, Schadendorf D, Artuc M, Grabbe J, and Henz BM (2000). Expression of SCF splice variants in human melanocytes and melanoma cell lines: Potential prognostic implications. Br J Cancer 82: 1453–1458.
Witte ON (1990). Steel locus defines new multipotent growth factor. Cell 63: 5–6.
Yamanaka Y, Friess H, Kobrin MS, Buchler M, Beger HG, and Korc M (1993). Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res 13: 565–569.
Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, and Ullrich A (1987). Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J 6: 3341–3351.
Zhang S, Anderson DF, Bradding P, Coward WR, Baddeley SM, MacLeod JD, McGill JI, Church MK, Holgate ST, and Roche WR (1998). Human mast cells express stem cell factor. J Pathol 186: 59–66.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Esposito, I., Kleeff, J., Bischoff, S. et al. The Stem Cell Factor–c-kit System and Mast Cells in Human Pancreatic Cancer. Lab Invest 82, 1481–1492 (2002). https://doi.org/10.1097/01.LAB.0000036875.21209.F9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000036875.21209.F9
This article is cited by
-
Prognostic and Clinic-Pathological Significances of SCF and COX-2 Expression in Inflammatory and Malignant Prostatic Lesions
Pathology & Oncology Research (2019)
-
Mechanisms of receptor tyrosine kinase activation in cancer
Molecular Cancer (2018)
-
Targeting kinases with anilinopyrimidines: discovery of N-phenyl-N’-[4-(pyrimidin-4-ylamino)phenyl]urea derivatives as selective inhibitors of class III receptor tyrosine kinase subfamily
Scientific Reports (2015)
-
A survival Kit for pancreatic beta cells: stem cell factor and c-Kit receptor tyrosine kinase
Diabetologia (2015)
-
The immune network in pancreatic cancer development and progression
Oncogene (2014)
