
Abstract
Chromosomal information on germ cell tumors of the infantile testis, ie, teratomas and yolk sac tumors, is limited and controversial. We studied two teratomas and four yolk sac tumors using comparative genomic hybridization (CGH) and in situ hybridization. No chromosomal anomalies were found in the teratomas by any of the methods, not even after CGH on microdissected tumor cells. All yolk sac tumors showed aneuploidy, loss of parts of 4q and 6q, and gain of parts of 20q. Underrepresentation of parts of 8q and overrepresentation of parts of 3p, 9q, 12p, 17, 19q, and 22 were detected in most cases. In addition, one recurrent yolk sac tumor after a sacral teratoma was studied, showing a highly similar pattern of imbalances. While CGH demonstrated loss of 1p36 in one testicular yolk sac tumor, in situ hybridization revealed loss of this region in all yolk sac tumors. High-level amplification of the 12q13-q14 region was found in one yolk sac tumor. MDM2, of which the encoding gene maps to this chromosomal region, was found in all cases using immunohistochemistry, whereas no p53 could be detected. Accordingly, no mutations within exons 5 to 8 of the p53 gene were observed. These data prove the absence of gross chromosomal aberrations in teratomas of the infantile testis and show a characteristic pattern of gains and losses in the yolk sac tumors. Besides confirmation of previously found anomalies, recurrent losses of 1p21–31 and 4q23–33 and gains of 9q34 and 12p12–13 have not been reported before. While genetic inactivation of p53 seems unimportant in the pathogenesis of these tumors, biochemical inactivation by MDM2 might be involved. These data support the existence of three entities of germ cell tumors of the human testis: teratomas and yolk sac tumors of infants, seminomas and nonseminomas of adolescents and young adults, and spermatocytic seminomas of the elderly, each with its own specific pathogenesis.
Similar content being viewed by others


Introduction
Human germ cell tumors comprise a heterogeneous group of neoplasms that can be found at different anatomical localizations (gonads and several extragonadal sites), and present clinically at different ages (Looijenga and Oosterhuis 1999, for review; Oosterhuis et al, 1997). Within the testis, three entities can be distinguished. The most common type are those of adolescents and young adults: the seminomas and nonseminomas (Mostofi et al, 1987). They originate from carcinoma in situ (CIS) (Skakkebæk, 1972), which can be frequently found in preserved parenchyma adjacent to an invasive tumor. Both seminomas and nonseminomas are characterized by gain of the complete short arm of chromosome 12 (Sandberg et al, 1996, for review). In addition, these tumors express wild type p53 extensively (Guillou et al, 1996; Schenkman et al, 1995). Less common are spermatocytic seminomas, mostly found in elderly men (Burke and Mostofi, 1993). We demonstrated recently using karyotyping and comparative genomic hybridization (CGH) that these tumors are characterized by gain of chromosome 9 (Rosenberg et al, 1998b). The third group of testicular germ cell tumors comprise those found at a young age, histologically divided into teratoma and yolk sac tumor, although other histological elements have sporadically been reported. In principle, teratomas can be found as part of the entity composed of teratomas/yolk sac tumors, and as part of nonseminomas (see above), which can be found at different anatomical localizations. In addition cystic teratomas, ie, dermoid cysts, can develop in the ovary. The developmental potential and known chromosomal constitution of these entities have been extensively discussed (Oosterhuis et al, 1997).
Information about the chromosomal constitution of germ cell tumors of infants, including those of the testis, is limited, mainly because of their rareness and occasional inconsistency. Flow cytometry has demonstrated that teratomas are generally diploid, whereas yolk sac tumors can be either diploid, tetraploid, or aneuploid (Kommoss et al, 1990; Silver et al, 1994). This difference was confirmed by karyotyping, which showed no aberrations in teratomas, whereas anomalies of the short arm of chromosome 1, in particular loss of band p36, the long arm of chromosome 6, and abnormalities of 3p were observed in yolk sac tumors (Bussey et al, 1999; Hoffner et al, 1994; Kaplan et al, 1979; Oosterhuis et al, 1988, 1993; Perlman et al, 1994; Stock et al, 1995, 1996). In situ hybridization (ISH) supported absence of numerical abnormalities in teratomas but demonstrated, in addition to chromosome 1 aberrations, also copy number changes for chromosomes 8, 10, 12, 17, and X in the yolk sac tumors (Hu et al, 1992; Jenderny et al, 1995; Perlman et al, 1996; Stock et al, 1994). The cytogenetic studies so far include only a few informative cases, possibly explained by a loss of tumor cells during sample preparation. Even the largest study on pediatric germ cell tumors (Bussey et al, 1999) reported only one testicular yolk sac tumor with an abnormal karyotype, in the case of a patient under the age of 10 years. Furthermore, conflicting data with respect to 12p (Jenderny et al, 1995; Perlman et al, 1994; Stock et al, 1995) and the presence of CIS (Hawkins et al, 1997; Jørgensen et al, 1995; Stamp and Jacobsen, 1993) have been found. These uncertainties fuel the discussion about the pathogenetic relationship between germ cell tumors of the infantile testis and seminomas and nonseminomas.
We performed ISH and CGH on a series of two teratomas and four yolk sac tumors of the infantile testis. Both teratomas were also karyotyped. In addition, a recurrent yolk sac tumor of a sacral teratoma was studied. CGH can be performed on fresh, frozen, and formalin-fixed (archival) material and allows a screen of chromosomal imbalances throughout the whole genome within a single experiment (Kallioniemi et al, 1992). This method was found to be highly informative in a previous study, even when applied on a relatively small number of cases (Rosenberg et al, 1998b). In case of an absence of imbalances, CGH was also performed with DNA from microdissected tumor cells. The role of p53 and MDM2 was investigated by immunohistochemistry and mutation analysis of exons 5 to 8 of p53.
Results
The age of the patients at clinical presentation was 10 months for both teratomas, and 16, 18, 21, and 24 months for the yolk sac tumors. Both teratomas presented at stage I, and were treated with surgery only. The patients were disease free 80 and 84 months after initial diagnosis. All testicular yolk sac tumors presented also at stage I. After surgical removal of the affected testis, the elevated alpha-fetoprotein level decreased according to half-life. Within several months, however, a gradually elevated alpha-fetoprotein level was observed in two patients (Cases 4 and 5), and multiple metastases in lung, abdomen, and lymph nodes, adjacent to the left kidney and the para-aortic region, were identified. Chemotherapeutic treatment resulted in complete remission, both by marker analysis and scanning techniques, although follow-up is less than 1.5 years. The other two patients (Cases 1 and 2) are still disease free 6 and 12 years after initial diagnosis. Testicular parenchyma could be observed in all cases in which immature germ cells, but no CIS, were found (not shown). In addition, one recurrent yolk sac tumor of a sacral teratoma (Case 3) was investigated. The teratoma was diagnosed at birth, whereas the yolk sac tumor presented 19 months later with multiple metastases in lymph nodes in the pelvic and spinal cord regions. After chemotherapy, a residual mature teratoma was found after surgical intervention. Twelve years after initial diagnosis, the patient is still disease free.
Classical karyotyping after short-term in vitro culturing showed 46,XY for both testicular teratomas. The sacral teratoma was previously found to be diploid, and aneuploidy was observed in the recurrent yolk sac tumor (Oosterhuis et al, 1993). None of the testicular yolk sac tumors were karyotyped successfully. No chromosomal aberrations were detected with CGH in the teratomas using DNA isolated from the total sample. Therefore, the tumor cells were specifically purified from frozen tissue sections using microdissection (Fig. 1A). DNA was isolated from these cells and used again for CGH (“Materials and Methods” section). Even with DNA isolated from these enriched tumor cells, no chromosomal imbalances were observed (not shown). As indicated previously (Rosenberg et al, 1998a; 2000), ISH with a specific probe against a chromosome showing a balanced pattern in CGH is useful when seeking information about the ploidy of the cells under investigation. ISH, using a chromosome 13-specific probe, revealed only tumor cells with two or fewer signals per interphase nucleus (mean, 1.8/sd 0.46 and 1.9/0.58, respectively, not shown), supporting diploidy. Because of the high percentage of tumor cells in the yolk sac tumors (> 80%) (Fig. 2, lower panel), no microdissection of the tumor cells and subsequent DNA amplification before CGH has been performed for these samples. All cases showed chromosomal imbalances, summarized in Figure 3A, of which corresponding profiles are shown in Figure 3B. Except for chromosome 13, all chromosomes showed imbalances in at least one of the samples. In particular, loss of parts of 4q and 6q and gain of parts of 20q were observed in all cases, and gain of parts of 3p, 9q, 12p (see below), 17, 19q, and 22, and loss of parts of 8q, was observed in the majority of them. The yolk sac tumors that developed distant metastases (Cases 4 and 5) showed specifically loss of parts of chromosome 5 and gain of parts of 14q and chromosome 21. Although the recurrent yolk sac tumor after a sacral teratoma (Case 3) showed a pattern of imbalances highly similar to that found in the primary testicular yolk sac tumors (Fig. 3A), loss of a part of 18q and gain of a part of 4p was specifically observed in this tumor. Using ISH with the chromosome 13-specific probe, all yolk sac tumors (except Case 2, of which no appropriate material was available) exhibited tumor cells with three or more signals (between 12% and 30%) for chromosome 13, supporting aneuploidy (not shown).

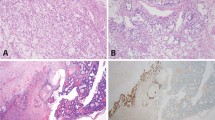
A, Representative example of a frozen tissue section (stained with hematoxylin and eosin) of a teratoma of the infantile testis, before (left panel) and after (right panel) microdissection. DNA was isolated and amplified prior to comparative genomic hybridization (CGH) as described in the “Materials and Methods” section. B, Representative example of the double fluorescent in situ hybridization, with one probe specific for the centromeric region of chromosome 1 (red signal) and another probe for the 1p36 region (green signal), on a formalin-fixed, paraffin-embedded yolk sac tumor of the infantile testis (Case 4). Note the reduced number of signals in the 1p36 region (green) compared with the centromere (red) (Table 1).

Representative examples of the immunohistochemical detection of the p53 protein (middle panel) and MDM2 protein (right panel) on a tissue section of a formalin-fixed, paraffin-embedded teratoma (upper panel) and a yolk sac tumor (lower panel) of the infantile testis. No p53 could be detected, whereas all tumor cells showed the presence of MDM2. The corresponding hematoxylin- and eosin-stained slide is represented on the left side of the illustration (magnification × 100).

A, Summary of the chromosomal imbalances detected by CGH in four yolk sac tumors of the infantile testis (1, 2, 4, and 5) and one recurrent yolk sac tumor of a sacral teratoma (3). DNA of all cases, except Case 4, was isolated from snap-frozen tissue. DNA of Case 4 was obtained from formalin-fixed, paraffin-embedded tissue. CGH was performed on DNA isolated directly from the samples, without amplification. Lines on the left and right side of the ideograms indicate under- and overrepresentation of chromosome material. The thick lines indicate the regions of high-level amplification. The numbers on top of each line refer to the case analyzed. B, Average profiles of Case 1 (see also panel A). Gains of parts of 3p, 9q,17, 19q, 20q, and 22, and losses of parts of 4 and 6, as well as the amplification of a discrete region on the long arm of chromosomes 12 and 17, can be seen.
CGH showed loss of the 1p21-p31 region in two testicular tumors and in the recurrent yolk sac tumors, whereas loss of 1p36 was found in only one testis. Double fluorescent ISH, using a probe for the centromeric region of chromosome 1 in combination with a 1p36-specific probe, showed significant loss of the 1p36 region in all yolk sac tumors but not in the teratomas (Table 1; Fig. 1B). This particular set of probes showed a 1:1 ratio in all controls included (not shown).
One testicular yolk sac tumor (Case 1) showed high-level amplification of the 12q13-q14 region, and although at a lower level, of region 17q12-q21 (Figure 3B). Immunohistochemistry showed that all teratomas and yolk sac tumors are positive for MDM2 (Figure 2), which the corresponding gene maps to 12q13-q14. No p53 protein could be observed (Figure 2), and no mutation within exons 5 to 8 of this gene were found (not shown).
Discussion
The pathogenetic relationship between the rare testicular germ cell tumors of infants and the more frequent testicular germ cell tumors of adolescents and young adults, ie, seminomas and nonseminomas, is a matter of debate. This is partly because of misdiagnosis of testicular germ cell tumors of adolescents and young adults as those of the infantile testis. In our study, the oldest patient was 24 months of age at clinical diagnosis, thereby excluding this possibility. Indeed, the histologies found in this series were only teratoma and yolk sac tumor, as previously suggested to occur in this particular subgroup (Oosterhuis et al, 1997). In addition, none of the cases showed CIS in the adjacent parenchyma, which are the precursor cells of seminomas and nonseminomas (Skakkebæk, 1972).
Information about the chromosomal constitution of germ cell tumors of the infantile testis is limited, and on some points, contradictory. Both flow cytometry and karyotyping suggested that teratomas of the infantile testis are diploid without gross chromosomal aberrations (Kaplan et al, 1979; Silver et al, 1994; Stock et al, 1995). To exclude misinterpretation caused by loss of tumor cells during sample preparation, we performed CGH and ISH. No chromosomal aberrations were observed, not even using DNA for CGH derived from microdissected tumor cells. These results prove for the first time that teratomas of the infantile testis are diploid, without gross numerical chromosomal anomalies. However, the teratomatous components of nonseminomas, as found in the adult testis, show the characteristic chromosomal aberrations present in other nonseminomatous elements of the adult testis, namely, eganeuploidy and gain of the short arm of chromosome 12 (Looijenga et al, 2000; Oosterhuis et al, 1997; van Echten et al, 1995).
In contrast, chromosomal aberrations were observed in the yolk sac tumors. In particular, loss of parts of 4q (23–33) and 6q (16–22) and gain of parts of 20q were found in all cases, and loss of parts of 1p (21–31) and 8q (23) and gain of parts of 3p (22–24), 9q (34), 12p (12–13), 17q (22–25), 19q (13), and 22 (13) were found in at least three cases (the minimal overlapping regions are indicated between brackets). These data provide a clear overview of the chromosomal imbalances most often found in yolk sac tumors of the infantile testis. Several of these aberrations are in accordance with earlier findings. In particular, loss of 6q and 3p aberrations have been consistently found in the few karyotyped cases (Bussey et al, 1999; Oosterhuis et al, 1988, 1993; Perlman et al, 1994). In addition, imbalances of (parts of) chromosome 8, 17, 19, 20, and 22, have been reported occasionally (Jenderny et al, 1995; Oosterhuis et al, 1988). Loss of the 1p36 region, consistently found in infantile yolk sac tumors, including those of the testis (Bussey et al, 1999; Jenderny et al, 1995; Perlman et al, 1994, 1996; Stock et al, 1995), is supported by our ISH data. However, the recurrent anomalies found on chromosomes 1p (21–31) and 4q (losses), and 9q and 12p (12–13) (gains), are novel. It should be noted that development of distant metastases might be related to loss of parts of chromosome 5 and gain of parts of 14q and 21. This deserves further investigation.
The recurrent yolk sac tumor of a sacral teratoma included in this study showed a highly similar pattern of imbalances compared with those of the infantile testis, in accordance with two cases studied by karyotyping (Perlman et al, 1994). This suggests that the chromosomal makeup of this tumor is more associated with its histology than with its anatomical site. We recently performed CGH on a pure yolk sac tumor component of a mixed nonseminoma of the adult testis (Looijenga et al, 2000). Although, in general, this yolk sac component showed a different pattern of chromosomal losses and gains compared with the results found in this study, loss of 6q was an exception. This anomaly was specifically found in the yolk sac tumor component of the nonseminoma of the adult testis, as well as in all yolk sac tumors included in this study, which suggests that it is related to yolk sac histology.
Three of the yolk sac tumors of the infantile testis showed overrepresentation of the 12p12–13 region, specifically, which might explain the inconsistency found in the literature (Jenderny et al, 1995; Perlman et al, 1994; Stock et al, 1995). Gain of the complete short arm of chromosome 12 is the most recurrent, and characteristic, chromosomal aberration in testicular germ cell tumors of adolescents and young adults (Sandberg et al, 1996, for review). In addition, high-level amplification of the 12p11.2–12.1 region can be found (Mostert et al, 1998). Moreover, both seminomas and nonseminomas are consistently aneuploid (Mostert et al, 1996; Oosterhuis et al, 1989; Rosenberg et al, 2000; van Echten et al, 1995), with a characteristic pattern of numerical aberrations: gain of parts of chromosomes 7, 8, 12 (short arm), and X, and loss of (parts of) 4, 5, 11, 13, 18, and Y. We also showed that gain of chromosome 9 is specifically found in spermatocytic seminomas (Rosenberg et al, 1998b). These patterns are clearly different from those found in the yolk sac tumors (and teratomas) of the infantile testis as reported here.
The available data so far favor independent pathogenetic pathways for germ cell tumors of the infantile testis and those of the adult testis, ie, seminomas, nonseminomas, and spermatocytic seminomas. This is also supported by the absence of p53, both in teratomas and yolk sac tumors, whereas it has been consistently found in seminomas and nonseminomas (Guillou et al, 1996; Schenkman et al, 1995). Because of this finding and the absence of mutations within exons 5 to 8 of this gene, genetic inactivation of p53 in infantile testicular germ cell tumors is unlikely, although presence of the MDM2 protein might inactivate p53 by proteolytic breakdown (Prives, 1998, for review). Amplification of the 12q13-q14 region, as found in one of the yolk sac tumors studied, and the immunohistochemical data are compatible with such an involvement of MDM2.
In conclusion, the three entities of testicular germ cell tumors, ie, teratomas and yolk sac tumors of infants, seminomas and nonseminomas of adolescents and young adults, and spermatocytic seminomas, have separate pathogenetic pathways that are associated with characteristic chromosomal anomalies. Application of ISH, with probes for the chromosomal regions specifically lost or gained in the different groups of testicular germ cell tumors, might be applicable in a diagnostic setting.
Materials and Methods
Samples
The samples included in this study were collected in close collaboration with urologists and pathologists in the South-Western part of the Netherlands and the University Hospital in Groningen. Directly after surgical removal, representative parts of the tumor and adjacent normal tissue (when available) were snap-frozen and other pieces were fixed overnight in 10% buffered formalin and subsequently embedded in paraffin. In one case (4), only paraffin-embedded material was available. The tumors were diagnosed according to the World Health Organization classification for testicular tumors (Mostofi, 1980). Two cases were pure mature teratomas, whereas four were pure yolk sac tumors. In addition, we studied one recurrent yolk sac tumor of a sacral teratoma described by us previously (Oosterhuis et al, 1993).
Metaphase Preparations
Representative parts of the tumor were enzymatically digested (collagenase; Sigma, St. Louis, Missouri) and cultured in T75 flasks (Costar, Cambridge, Massachusetts) for several days under standard conditions, ie, 37° C in a humidified atmosphere of 5% CO2 in DMEM/HF12 with 10% heat-inactivated FCS (BRL-GIBCO, Gaithersburg, Maryland), as reported previously (Castedo et al 1989). Mitotic cells were harvested after 2 to 4 hours of colcemid treatment, swollen in hypotonic solution, and fixed in methanol:acetic acid (3:1). For conventional G-band karyotyping, air-dried preparations were digested with pancreatin (Sigma). The chromosome constitution was described according to the International System for Human Cytogenetic Nomenclature 1995 (Mitelman, 1995). For CGH, metaphase spreads were prepared using standard procedures from in vitro phytohemagglutinin-stimulated peripheral blood lymphocyte cultures of a healthy male.
Comparative Genomic Hybridization
High molecular weight DNA was isolated from the snap-frozen tissue samples (test DNA) and from peripheral blood of a normal male (reference DNA) using standard procedures (Maniatis et al, 1982). DNA of the paraffin-embedded yolk sac tumor was isolated as described before (Alers et al, 1997). In case no aberrations were identified on the initial DNA sample (both teratomas), DNA of purified tumor cells was acquired from frozen tissue as follows. Tissue sections (20-ìm thick) of the samples were cut and air dried. In addition, a hematoxylin and eosin staining of a parallel tissue section (4-ìm thick) was performed for additional histological control. The cells of interest were identified using an inverted microscope (Axiovert 10; Carl Zeiss, Jena, Germany) and microdissected with a self-made glass capillary piece. The section after microdissection was also stained for histological check. The tissue parts were put directly into a reaction vessel and dissolved in TNE (Tris, 10 mm; NaCl, 400 mm; EDTA, 2 mm, pH 8,2). Proteinase K (10 mg/ml) and SDS (10%) were added, and incubation was done overnight at 37° C. Subsequently, DNA was extracted with phenol/chloroform/isoamylalcohol (25:24:1) (Merck, Darmstadt, Germany), and precipitation was performed with glycogen (10 mg/ml) (Boehringer, Mannheim, Germany) as carrier. Pellets were dissolved in TE (Tris, 10 mm; EDTA 0, 1 mm, pH 8,0). Eight to 25 ng of DNA from each microdissected teratoma sample and from a normal male were amplified by standard degenerate oligonucleotide primed polymerase chain reaction (DOP-PCR) (Telenius et al, 1992). The quality of this approach was checked with control DNA amplified with the same procedure derived from normal males (XY) and females (XX). A total of seven independent experiments were performed, and all showed a balanced profile for the autosomes. The sex chromosomes showed the expected imbalances in gender-mismatched hybridizations. In addition, CGH was performed on genomic DNA from an osteosarcoma of a patient with Rothmund-Thomson syndrome, both before and after DOP-PCR. From the 20 imbalances found in the genomic DNA, 18 were observed after amplification (two false negatives). No aberrations were detected in the sample after amplification (no false positives). This method was reproducibly applicable on 8 ng DNA, equivalent to approximately 900 nuclei with a diploid DNA content (not shown). Of the samples studied, DNA equivalent of at least 1500 nuclei was used for the amplification. This approach was successfully used to investigate the chromosomal imbalances of various developmental stages of both seminoma and nonseminoma, including CIS (Looijenga et al, 2000).
The CGH procedure and analysis was performed as described previously (Rosenberg et al, 1998a; 1998b). Both the control male DNA and the tumor DNA were directly labeled by nick-translation with lissamine and FITC respectively. The data were analyzed using Quips XL software from Vysis (Downers Grove, Illinois). Normalization was carried out using the average of the green to red fluorescent intensities for the entire metaphase. Five metaphases were studied for each case. Losses of DNA sequences were defined as chromosomal regions where the average green-to-red ratio and its 95% confidence interval are below 0.9, whereas gains are above 1.1. These narrow thresholds of 0.9 and 1.1 are suitable for the direct labeling procedure, as used in this study (see above) and as discussed by us previously (Rosenberg et al, 1998c). The results obtained, with and without DOP-amplification, resulted in narrow 95% confidence intervals (not shown and Looijenga et al, 2000). The heterochromatic blocks of chromosome 1, 9, 16, and Y, and the immediate telomeric regions were excluded from the analysis because these regions present variable results in experiments with normal control DNAs.
In Situ Hybridization
ISH was performed with a chromosome 13 probe (YAC 908C2, mapped to 13q34) (Kingsley et al, 1997) kindly provided by B. Eussen (Department of Clinical Genetics, Erasmus University, Rotterdam, The Netherlands). Because chromosome 13 did not show imbalances in any of the tumors by CGH, combination of the ISH and CGH data made discrimination between diploid and polyploidy possible. This approach has been found to be informative in previous studies of a cell line (Rosenberg et al, 1998a) and of invasive seminomas and nonseminomas (Rosenberg et al, 2000).
Chromosome 1 band p36 is a critical region for CGH. Therefore, possible loss of the 1p36 region in addition to CGH was studied in more detail by means of a double fluorescent ISH approach using one probe specific for the centromeric region of chromosome 1 (Cook and Hindley, 1979) and another probe specific for the 1p36 region (cosmid: D1S32, pBG2.8, kindly provided by Prof. dr.A. de Paepe, Centre of Medical Genetics, University of Gent, Belgium). Representative formalin-fixed, paraffin-embedded tissue sections of 5-μm thickness were used. The procedure used is a slightly modified method described previously (Hopman et al, 1992). Briefly, the sections were deparaffinized by xylene and methanol and pretreated in 1M NaSCN at 80° C and pepsin (8000 U/ml) at 37° C, respectively, after which they were rinsed in distilled water and PBS and dehydrated. Probes were labeled by nick-translation, according to standard procedures, either with dioxigenin-11-dUTP (Boehringer) or biotin-16-dUTP, and applied in 10 to 15 μl hybridization mixture on the tissue slides (13q probe, 20 ng/μl; 1p36, 2 ng/μl; chromosome 1 centromere, 2 ng/μl). The probes were denatured together with the target by placing the slide for 10 minutes on the bottom of a 80° C oven. After hybridization overnight at 37° C, the slides were washed stringently, and the hybrids were detected by FITC-conjugated sheep-antidigoxigenin (Boehringer) and CY3-conjugated avidine (Jackson Immuno Research Laboratories, Inc., Westgrove, Pennsylvania) as described by us previously (Mostert et al, 1998). Results were studied with a Zeiss Axioskop fluorescence microscope (Carl Zeiss) with a Pinkel filter in combination with a triple band-pass filter to visualise FITC, CY3, and DAPI simultaneously. This set of probes was verified to be useful on a series of normal tissues, showing a 1:1 ratio (not shown), as well as on host cells within the same histological section in which the tumor cells were investigated. The tumor component was only scored when a 1:1 ratio was found in the controls. A total of 30 nuclei of each component were investigated, among which possible differences were investigated using Student's t test analysis.
p53 Mutation Analysis
Because the majority (78%) of the missense mutations in the p53 gene are found in exons 5 to 8, the tumors were screened for mutations in this region using PCR amplification and subsequent single strand conformation polymorphism analysis (SSCP), as described previously (Schuyer et al, 1998). Briefly, the specific exon regions were amplified using commercially available primers (Clontech, Palo Alto, California) and analyzed by single strand conformation polymorphism analysis. PCR products showing an altered electrophoretic mobility were re-amplified and sequenced by double stranded sequencing.
Immunohistochemistry
Immunohistochemistry was performed on representative formalin-fixed, paraffin-embedded tissue sections (3-μm thickness) of both teratomas and all yolk sac tumors. The p53-specific antibodies (DO-7; DAKO A/S, Glostrup, Denmark) were used in a final concentration of 80 μg/ml, and the MDM2-specific antibodies (clone SMP14; NeoMarkers, Union City, New Jersey) in a final concentration of 40 μg/ml. The sections were pretreated with a modified heath-induced antigen retrieval method (Shi et al, 1991) using 10 mmol/l citrate buffer, pH 6.0. The detection was done with the avidin-biotin-peroxidase complex (DAKO A/S) according to the manufacturer's instructions using 3,3′-diaminobenzidine as chromogen. Sections were lightly counterstained with hematoxylin. In all experiments, both positive (samples with known presence of p53 and MDM2) and negative controls (samples without the presence of p53 and MDM2 and without the first antibody) were included. Photographs were made using an Olympus BX 50 microscope, equipped with a Olympus DP 10 camera and the Olympus DP-Soft analysis system version 3.0 for Windows (Olympus, Hamburg, Germany).
References
Alers JC, Krijtenburg PJ, Rosenberg C, Hop WC, Verkerk AM, Schröder FH, Van der Kwast TH, Bosman FT, and Van Dekken H (1997). Interphase cytogenetics of prostatic tumor progression: Specific chromosomal abnormalities are involved in metastasis to the bone. Lab Invest 77:437–448.
Burke AP and Mostofi FK (1993). Spermatocytic seminoma. A clinicopathologic study of 79 cases. J Urol Pathol 1:21–32.
Bussey KJ, Lawce HJ, Olson SB, Arthur DC, Kalousek DK, Krailo M, Giller R, Heifetz S, Womer R, and Magenis RE (1999). Chromosome abnormalities of eighty-one pediatric germ cell tumors: Sex-, age-, site-, and histopathology-related differences: A Children's Cancer Group study. Genes Chromosomes Cancer 25:134–146.
Castedo SM, De Jong B, Oosterhuis JW, Seruca R, Idenburg VJS, Dam A, Te Meerman GJ, Koops H, Sleijfer DT (1989). Chromosomal changes in human primary testicular nonseminomatous germ cell tumors. Cancer Res 49:5696–5701.
Cook HJ and Hindley J (1979). Cloning of human alpha satellite III DNA: Different components are on different chromosomes. Nucleic Acids Res 6:3177–3197.
Guillou L, Estreicher A, Chaubert P, Hurlimann J, Kurt AM, Metthez G, Iggo R, Gray AC, Jichlinski P, and Benhattar J (1996). Germ cell tumors of the testis overexpress wild-type p53. Am J Pathol 149:1221–1228.
Hawkins E, Heifetz SA, Giller R, and Cushing B (1997). The prepubertal testis (prenatal and postnatal): Its relationship to intratubular germ cell neoplasia: A combined Pediatric Oncology Group and Children's Cancer Study Group. Hum Pathol 28:404–410.
Hoffner L, Deka R, Chakravarti A, and Surti U (1994). Cytogenetics and origins of pediatric germ cell tumors. Cancer Genet Cytogenet 74:54–58.
Hopman AHN, Poddigde P, Moesker O, and Ramaekers FCS (1992). Interphase cytogenetics: An approach to the detection of genetic aberrations in tumours. In: Herrington CS and McGee JOD, editors. Diagnostic molecular pathology: A practical approach. Oxford, UK: IRL Press, 141–163.
Hu LM, Phillipson J, and Barskym SH (1992). Intratubular germ cell neoplasia in infantile yolk sac tumor. Verification by tandem repeat sequence in situ hybridization. Diagn Mol Pathol 1:118–128.
Jørgensen N, Muller J, Giwercman A, Visfeldt J, Moller H, and Skakkebæk NE (1995). DNA content and expression of tumour markers in germ cells adjacent to germ cell tumours in childhood: Probably a different origin for infantile and adolescent germ cell tumours. J Pathol 176:269–278.
Jenderny J, Koester E, Meyer A, Borchers O, Grote W, Harms D, and Jaenig U (1995). Detection of chromosome aberrations in paraffin sections of seven gonadal yolk sac tumors of childhood. Hum Genet 96:644–650.
Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, and Pinkel D (1992). Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258:818–820.
Kaplan CG, Askin FB, and Benirschke K (1979). Cytogenetics of extragonadal tumors. Teratology 19:261–266.
Kingsley K, Wirth J, van der Maarel S, Freier S, Ropers HH, and Haaf T (1997). Complex FISH probes for the subtelomeric regions of all human chromosomes: Comparative hybridization of CEPH YACs to chromosomes of the Old World monkey Presbytis cristata and great apes. Cytogenet Cell Genet 78:12–19.
Kommoss F, Bibbo M, and Talerman A (1990). Nuclear deoxyribonucleic acid content (ploidy) of endodermal sinus (yolk sac) tumor. Lab Invest 62:223–231.
Looijenga LHJ and Oosterhuis JW (1999). Pathogenesis of testicular germ cell tumors. Rev Reprod 4:90–100.
Looijenga LHJ, Rosenberg C, Van Gurp RJHLM, Geelen E, van Echten-Arends J, De Jong B, Mostert MC, and JW Oosterhuis (in press, 2000). Comparative genomic hybridization of microdissected samples from different stages in the development of a seminoma and nonseminoma. J Pathol.
Maniatis T, Fritsch EF, and Sambrook J (1982). Isolation of high molecular-weight, eukaryotic DNA from cells grown in tissue culture. In: Molecular cloning. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory, 280–281.
Mitelman F (1995). ISCN 1995: An international system for human cytogenetic nomenclature. Basel, Switzerland: S. Karger.
Mostert MC, Verkerk AJ, Van de Pol M, Heighway J, Marynen P, Rosenberg C, van Kessel A, van Echten J, Oosterhuis JW, and Looijenga LHJ (1998). Identification of the crucial region of 12p overrepresentation in testicular germ cell tumors of adolescents and adults. Oncogene 16:2617–2627.
Mostert MM, Van de Pol M, Olde Weghuis D, Suijkerbuijk RF, Geurts van Kessel A, van Echten-Arends J, Oosterhuis JW, and Looijenga LHJ (1996). Comparative genomic hybridization of germ cell tumors of the adult testis: Confirmation of karyotypic findings and identification of a 12p-amplicon. Cancer Genet Cytogenet 89:146–152.
Mostofi FK (1980). Pathology of germ cell tumors of testis. A progress report. Cancer 45:1735–1754.
Mostofi FK, Sesterhenn IA, and Davis CJJ (1987). Immunopathology of germ cell tumors of the testis. Semin Diagn Pathol 4:320–341.
Oosterhuis JW, Castedo SM, de Jong B, Cornelisse CJ, Dam A, Sleijfer DT, and Schraffordt Koops H (1989). Ploidy of primary germ cell tumors of the testis. Pathogenetic and clinical relevance. Lab Invest 60:14–20.
Oosterhuis JW, Castedo SM, de Jong B, Seruca R, Buist J, Schraffordt Koops H, and Leeuw JB (1988). Karyotyping and DNA flow cytometry of an orchidoblastoma. Cancer Genet Cytogenet 36:7–11.
Oosterhuis JW, Looijenga LHJ, van Echten-Arends J, and De Jong B (1997). Chromosomal constitution and developmental potential of human germ cell tumors and teratomas. Cancer Genet Cytogenet 95:96–102.
Oosterhuis JW, Van Berlo RJ, de Jong B, Dam A, Buist J, Tamminga RYJ, and Zwierstra RP (1993). Sacral teratoma with late recurrence of yolk sac tumor: Human counterpart of embryo- or yolk sac-derived teratoma? J Urol Pathol 1:257–267.
Perlman EJ, Cushing B, Hawkins E, and Griffin CA (1994). Cytogenetic analysis of childhood endodermal sinus tumors: A Pediatric Oncology Group study. Pediatr Pathol 14:695–708.
Perlman EJ, Valentine MB, Griffin CA, and Look AT (1996). Deletion of 1p36 in childhood endodermal sinus tumors by two-color fluorescence in situ hybridization: A pediatric oncology group study. Genes Chromosomes Cancer 16:15–20.
Prives C (1998). Signaling to p53: Breaking the MDM2–p53 circuit. Cell 95:5–8.
Rosenberg C, Bakker-Schut T, Mostert MC, Tanke H, Raap AK, Oosterhuis JW, and Looijenga LHJ (1998a). Comparative genomic hybridisation in hypotriploid/hypertriploid tumors. Cytometry 29:113–121.
Rosenberg C, Mostert MC, Bakker Schut T, Van de Pol M, van Echten-Arends J, De Jong B, Raap T, Tanke H, Oosterhuis JW, and Looijenga LHJ (1998b). Chromosomal constitution of human spermatocytic seminomas: Comparative genomic hybridization supported by conventional and interphase cytogenetics. Genes Chromosomes Cancer 23:286–291.
Rosenberg C, Van Gijlswijk RP, Vos CBJ, Wiegant J, Cornelisse CJ, Tanke HJ, and Raap AK (1998c). Comparative genomic hybridization with lissamine- and fluorescein-labeled nucleotides. Cytometry 32:337–341.
Rosenberg C, Bakker Schut T, Mostert MC, Tanke H, Raap AK, Oosterhuis JW, and Looijenga LHJ (2000). Chromosomal gains and losses in testicular germ cell tumors of adolescents and adults investigated by a modified comparative genomic hybridization approach. Lab Invest 79:1447–1451.
Sandberg AA, Meloni AM, and Suijkerbuijk RF (1996). Reviews of chromosome studies in urological tumors. III. Cytogenetics and genes in testicular tumors. J Urol 155:1531–1556.
Schenkman NS, Sesterhenn IA, Washington L, Tong YA, Weghorst CM, Buzard GS, Srivastava S, and Moul JW (1995). Increased p53 protein does not correlate to p53 gene mutations in microdissected human testicular germ cell tumors. J Urol 154:617–621.
Schuyer M, Henzen-Logmans SC, van der Burg ME, Fieret EJ, Klijn JG, Foekens JA, and Berns EM (1998). High prevalence of codon 213Arg–>Stop mutations of the Tp53 gene in human ovarian cancer in the southwestern part of The Netherlands. Int J Cancer 76:299–303.
Shi SR, Key ME, and Kalra KL (1991). Antigen retrieval in formalin-fixed, paraffin-embedded tissues: An enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem 39:741–748.
Silver SA, Wiley JM, and Perlman EJ (1994). DNA ploidy analysis of pediatric germ cell tumors. Mod Pathol 7:951–956.
Skakkebæk NE (1972). Possible carcinoma-in-situ of the testis. Lancet 2:516–517.
Stamp IM and Jacobsen GK (1993). Intratubular germ cell neoplasia in an infantile testis. Histopathology 23:99–100.
Stock C, Ambros IM, Lion T, Haas OA, Zoubek A, Gadner H, and Ambros PF (1994). Detection of numerical and structural chromosome abnormalities in pediatric germ cell tumors by means of interphase cytogenetics. Genes Chromosomes Cancer 11:40–50.
Stock C, Ambros IM, Sthehl S, Zubek A, Fink FM, Gadner H, and Ambros PF (1995). Cytogenetic aspects of pediatric germ cell tumors. Klin Padiatr 207:235–241.
Stock C, Strehl S, Fink FM, Bauer S, Lion T, Kreczy A, Gadner H, and Ambros PF (1996). Isochromosome 12p and maternal loss of 1p36 in a pediatric testicular germ cell tumor. Cancer Genet Cytogenet 91:95–100.
Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, and Tunnacliffe A (1992). Degenerate oligonucleotide-primed PCR: General amplification of target DNA by a single degenerate primer. Genomics 13:718–725.
van Echten J, Oosterhuis JW, Looijenga LH, van de Pol M, Wiersma J, Te Meerman G, Schraffordt Koops H, Sleijfer DT, and De Jong B (1995). No recurrent structural abnormalities apart from i(12p) in primary germ cell tumors of the adult testis. Genes Chromosomes Cancer 14:133–144.
Acknowledgements
We acknowledge the pathologists and urologists in the South-Western part of the Netherlands for their help in collecting the samples. Mr. Ad J.M. Gillis and Mr. Ruud J.H.L.M. van Gurp are thanked for their technical support, and Dr. Janneke Alers (Pathology, Erasmus University, Rotterdam) for her help in generating the photographs of the in situ hybridization.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is supported by a grant from the Dutch Cancer Society (DDHK 94 836) Postgraduate School Molecular Medicine: Pathophysiology of Growth and Differentiation.
Rights and permissions
About this article
Cite this article
Mostert, M., Rosenberg, C., Stoop, H. et al. Comparative Genomic and In Situ Hybridization of Germ Cell Tumors of the Infantile Testis. Lab Invest 80, 1055–1064 (2000). https://doi.org/10.1038/labinvest.3780110
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780110
This article is cited by
-
Molekularpathologie bei urologischen Tumoren
Der Pathologe (2021)
-
Testicular cancer
Nature Reviews Disease Primers (2018)
-
Lessons from human teratomas to guide development of safe stem cell therapies
Nature Biotechnology (2012)
-
Methylator phenotype of malignant germ cell tumours in children identifies strong candidates for chemotherapy resistance
British Journal of Cancer (2011)
