
Abstract
Malignant fibrous histiocytoma (MFH) is the most common soft tissue sarcoma. Nevertheless, the validity of this heterogeneous pathological entity has been recurrently questioned by pathologists. Recently, analyses by comparative genomic hybridization (CGH) of a large series of MFHs suggested that MFHs consist of morphologic modulation of other poorly differentiated sarcomas like leiomyosarcomas (LMS) or dedifferentiated liposarcomas (DLPS). We report here an analysis by CGH of 22 myxoid MFHs (mMFH), one of the five histological subtypes of MFH, and of nine pleomorphic liposarcomas (pLPS), a rare poorly differentiated LPS. The chromosome imbalances encountered in the series of mMFH were very similar to those observed in the series of pLPS studied in the laboratory and in the series of 14 pLPS published in the literature. The most frequent gains involved chromosome subregions: pericentromeric regions of 1, 5p, 19p, 19q and 20q. Losses found in the chromosomal arms 1q, 2q, 3p, 4q, 10q, 11q and 13q were also recurrent. The use of a clustering software did not separate the two pathological groups (mMFH and pLPS) on the basis of genomic data. Moreover, pLPS–mMFH represented, according to the clustering software results, an entity clearly distinguished from other soft tissue sarcomas, LMS in particular, with which they share common genetic aberrations. Additional studies are needed to identify genes targeted by these genomic aberrations, and implicated in the oncogenesis of these tumor subtypes. The characterization of common gene alterations in both tumor groups would suggest a closer relationship between these two types of soft tissue sarcomas.
Similar content being viewed by others
Main
Malignant fibrous histiocytoma (MFH) is the most frequent soft tissue sarcoma of later adult life. Several clinicopathologic variants were described: the storiform pleomorphic form, the myxoid form, the giant-type cell form and the inflammatory-type form. Another group, the angiomatoid variant, has been excluded from soft tissue sarcomas and reclassified within the fibrohistiocytic tumors of low malignant potential. The clinicopathologic group of MFH is heterogeneous, and its validity and existence are often discussed.1, 2, 3 Comparative genomic hybridization (CGH) analyses of large series of MFHs have recently demonstrated that most MFHs do not constitute a homogeneous entity but could correspond to morphologic modulations in the tumoral progression of other sarcomas, particularly leiomyosarcoma (LMS)4 or liposarcoma (LPS).5
A preliminary study, by CGH, of a small series of myxoid MFHs (mMFH) revealed recurrent chromosome imbalances. The most frequent gains involved chromosomal arms 1p, 1q, 5p, 19q and 20q. Loss of the long arm of chromosome 13 was also recurrent. This genomic profile was also reported in a rare subtype of LPS, the pleomorphic liposarcoma (pLPS).6 Moreover, pathological features of pLPS are characterized by adipoblastic areas, which permit the diagnosis of lipomatous/adipose lineage tumor, but equally by areas devoid of adipoblasts, mimicking mMFH pathological features.7, 8 All these observations suggest that pLPS and mMFH could be closely related tumor subtypes.
In order to document these observations, we have studied a larger series of mMFH and pLPS, by CGH and clustering software.
Materials and methods
Tumor Analysis
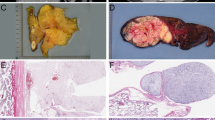
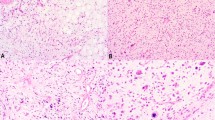
The samples were obtained from different pathology laboratories. A total of 30 tumors were obtained as fresh samples and one tumor as a cell line. All of the histological slides were re-examined by the French Sarcoma Group of the Fédération Nationale des Centres de Lutte Contre Le Cancer (FNCLCC): 22 tumors were classified as mMFH and nine as pLPS. According to the recommendations of the FNCLCC, the grading was established for each of the 14 primary tumors (from I to III). Two of the primary tumors were grade I, five were grade II and five were grade III. For two of them, data were not available. The main clinical and histopathological data are listed in Table 1.
DNA was extracted from all the samples using a phenol–chloroform procedure.
CGH and Image Analysis
CGH has been described previously.9 Tumor DNAs were directly labeled by nick-translation using an (FITC)-dUTP nucleotide (DuPont NEN, Boston, MA, USA). DNA fragments obtained ranged from 500 to 2000?bp. Control DNA was Spectrum Red normal male reference DNA (Vysis, Downers Grove, IL, USA). A volume of 1?μg of tumor DNA, 1?μg of control DNA and 80?μg of Cot-1 DNA (Gibco/BRL, Life Technologies, Gaithersburg, MD, USA) were mixed in 12?μl of hybridization buffer (formamide 50%, NaH2PO4 40?mM, SDS 0.1%, dextran sulfate 10%, 2 × SSC).
A Leica DMRB fluorescence microscope and a Photometrics Nu200 CCD camera were used to capture CGH pictures from 10 metaphases as a mean. Mitoses were analyzed with the Quips™ XL Software (Vysis, Downers Grove, IL, USA).
Green-to-red ratios greater than 1.2 and less than 0.8 were considered as gains and losses, respectively. Ratios greater than 1.4 were considered as high-level amplification and ratios greater than 1.6 as very high-level amplification. We have only taken into consideration experiments fulfilling the quality criteria previously defined.10 GC-rich chromosome regions, which are known to give false-positive results, were interpreted with caution, in particular for the low level of imbalances.
Clustering of the CGH Data and Classifying Algorithms
In order to compare genomic profiles of analyzed tumors, we used the clustering software Molmime J-Express (freely available for academic laboratories from http://www.molmime.com).
As previously described, we divided the whole karyotype into 150 digitized regions with a mean length of 20?Mb, a size close to the resolution obtained by CGH.5 The status amplified, loss or normal, of each region for a given tumor was arbitrarily fixed as follows: loss=−1, normal status=0, simple gain=+1, high amplification=+3 and very high amplification=+6. All the data obtained for the 150 regions and for all analyzed tumors were automatically loaded into a File Maker Pro format and then exported to the clustering software Molmime J-Express. The clustering software then classify the tumors that have similar genomic profiles, thus enabling easier comparison between tumors. The average linkage cluster method (Pearson correlation) was used.
Results
Imbalances Detected by CGH
All tumors investigated exhibited imbalances. The regions of DNA copy number imbalances are shown in Figure 1.

Ideogram of regions of DNA copy number imbalances. Vertical lines on the right and left sides of chromosomes indicate gains and losses, respectively. Color code of tumors: pLPS studied in the laboratory (light green), mMFH studied in the laboratory (blue).
Gains of DNA: Recurrent high-level DNA amplification (green-to-red ratio greater than 1.4) was observed in three different regions: central region of chromosome 1 in 20/31 tumors (14 mMFH and six pLPS), 5p in 17/31 tumors (11 mMFH and six pLPS) and 20q in 15/31 tumors (12 mMFH and three pLPS). Among these three chromosome regions, two of them are very highly amplified (green-to-red ratio greater than 1.6): 5p in 2/31 tumors (two pLPS) and 20q in one mMFH. Two chromosome subregions were frequently concerned with simple gains: 19p in 18 tumors (17 mMFH and one pLPS) and 19q in 17 tumors (16 mMFH and one pLPS). Other sites were less frequently (less than 50%) concerned with genomic gain.
Losses of DNA: As shown in Figure 1, losses of chromosome 13 were observed in 30 of the 31 tumors (22 mMFH and eight pLPS). Losses of 1q in 23 tumors (20 mMFH and three pLPS), 2q in 24 tumors (17 mMFH and seven pLPS), 3p in 17 tumors (13 mMFH and four pLPS), 4q in 17 tumors (14 mMFH and three pLPS), 10q in 17 tumors (12 mMFH and five pLPS) and 11q in 19 tumors (11 mMFH and eight pLPS) were also recurrent.
Genomic imbalances were observed in mMFH according to the grade. Data were available for nine mMFH (two grade 1, three grade 2 and four grade 3). Among recurrent chromosome imbalances observed in mMFH (see above), a gain of 5p and loss of 4q were not observed in 2/2 mMFH of grade 1 whereas both aberrations were detected in 4/7 of higher grade mMFH (Table 2).
Clustering
Comparison between 9 pLPS and 22 mMFH studied in the laboratory and 14 pLPS reported in literature
In order to compare the complex genomic imbalances observed in the group of pLPS and the group of mMFH, we pooled the CGH data of pLPS and mMFH of our series and those of pLPS published in the literature.6 No clear clustering was obtained, and the three series were completely interspersed all along the dendrogram (Figure 2). The clustering software was thus unable to separate the two pathologic groups (mMFH and pLPS). Moreover, the clustering software could not separate the groups of pLPS from the literature and from our series, suggesting that both groups share common genetics and thus correspond to a homogeneous genomic entity, authorizing us to pool these two groups of pLPS to have a more representative series.

Dendrogram of the CGH data obtained on 45 mMFH-pLPS. Each row corresponds to a tumor and each column to a chromosome region of 20?Mb. Code of tumors appears on the right and is associated with a color: LPSp reported in the literature (dark green), LPSp studied in the laboratory (light green), mMFH studied in the laboratory (blue). In a given row, 1p is on the left and 22q on the right. Amplification appears as yellow squares, gains as more or less green squares and losses as red squares. No discrimination is obtained between the two pathological groups that appear interspersed along the dendrogram.
Comparison between the group of 14 pLPS reported in the literature, 22 mMFH, nine pLPS, 35 LMS and five dedifferentiated liposarcomas studied in the laboratory
Based on CGH data, the clustering software distinguished three genomic entities: the group of pLPS–mMFH, the group of LMS and the group of dedifferentiated liposarcomas (DLPS). The group of pLPS–mMFH appeared as a distinct genomic entity different from the other two groups of soft tissue sarcomas (DLPS and LMS). However, pLPS–mMFH present some genomic imbalances very similar to those observed in LMS, in particular, loss of chromosome 13 (32/35), 1q (18/35), 2q (19/35), 4q (14/35), 10q (22/35) and 11q (21/35), and gain of 1p (20/35) and 19p (18/35) and 19q (15/35). Seven tumors (8% of the 85 tumors) did not respect this classification: four LMS (R4, R9, R17 and R27) and three mMFH (B6, B18 and B20) (Figure 3).

Dendrogram of the CGH data obtained on 45 mMFH-pLPS, and on 35 LMS and five DLPS. Each row corresponds to a tumor and each column to a chromosome region of 20?Mb. Color code of tumors appears on the right: pLPS studied in the laboratory (light green), pLPS reported in the literature (dark green), mMFH studied in the laboratory (blue), LMS studied in the laboratory (red), DLPS studied in the laboratory (yellow). In a given row, 1p is on the left and 22q on the right. Amplification appears as yellow squares, gains as more or less green squares and losses as red squares. Based on the CGH data, the clustering software distinguished three genomic entities: the group of pLPS-mMFH, the group of LMS and the group of DLPS.
Discussion
mMFH and pLPS, which were considered until now as two clinicopathologically distinct entities, are characterized by complex genomic profiles.
Some of the chromosome imbalances observed in the groups of pLPS and of mMFH are very similar to those observed in LMS, suggesting partially common genetics and biology.
Nevertheless, pLPS and mMFH present characteristic chromosome imbalances, which permit one to distinguish them clearly from LMS.
Genomic profiles of mMFH have never been reported in the literature. This study describes the genomic imbalances encountered in this pathologic group. The recurrent simple gains concerned the chromosome regions 19p and 19q. High-level amplifications of the central regions of chromosome 1, 5p and 20q were frequently observed. More rarely, very high-level amplifications were also observed in chromosome arms 5p and 20q. These highly amplified chromosome regions thus probably contain genes implicated in the oncogenesis and/or progression of this tumor group. The loss of chromosome 13 with a putative common region in band 13q14–21 is observed in 22/22 mMFH. We have previously demonstrated that the RB1 gene is the target of chromosome 13 deletions in pleomorphic MFH.11 Chromosome 13 losses observed in the present study could thus involve the RB1 locus.
Among the most characteristic chromosome imbalances observed in mMFH, the gain of 5p and loss of 4q are not observed in tumors of grade 1 whereas they are present in tumors of higher grades. This result, which must be confirmed in larger series, suggests that the gain of 5p and loss of 4q are probably late events in the oncogenesis of mMFH.
pLPS is a rare tumor representing about 1% of soft tissue malignancy. Only one CGH study is reported in the literature.6 Compared to our series of nine pLPS, the series of 14 specimens from the literature share the same chromosome imbalances, and the clustering software is unable to separate these two series of pLPS. This result permitted us to pool these two series in order to obtain a more representative group. The genomic imbalances encountered in the group of 23 pLPS were very similar to those observed in the group of 22 mMFH.
The clustering software suggests that the genomic profiles of pLPS and mMFH are closely related based on CGH data. Moreover, the comparison between the groups of mMFH–pLPS and two other groups of soft tissue sarcomas, LMS and DLPS, showed that the group of LPSp–MFHm is distinguished from the two other groups of soft tissue sarcomas. Only 7/85 tumors do not fit this classification. They could correspond to misclassified sarcomas.
CGH data and clustering software analysis show that these two groups share very similar complex chromosome imbalances. The most recurrent of them probably contain genes implicated in the oncogenesis of these tumors. Additional studies using other genetic techniques are warranted to analyze the possibility of a closer relationship between these two types of soft tissue sarcomas and to characterize gene alterations implicated in both tumors.
References
Fletcher CD . Malignant fibrous histiocytoma? Histopathology 1987;11:433–437.
Dehner LP . Malignant fibrous histiocytoma. Nonspecific morphologic pattern, specific pathologic entity, or both? Arch Pathol Lab Med 1988;112:236–237.
Fletcher CD, Gustafson P, Rydholm A, et al. Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: prognostic relevance of subclassification. J Clin Oncol 2001;19:3045–3050.
Derré J, Lagacé R, Nicolas A, et al. Leiomyosarcomas and most malignant fibrous histiocytomas share very similar comparative genomic hybridization imbalances: an analysis of a series of 27 leiomyosarcomas. Lab Invest 2001;81:211–215.
Chibon F, Mariani O, Mairal A, et al. The use of clustering software for the classification of comparative genomic hybridization data. An analysis of 109 malignant fibrous histiocytomas. Cancer Genet Cytogenet 2003;141:75–78.
Rieker RJ, Joos S, Bartsch C, et al. Distinct chromosomal imbalances in pleomorphic and in high-grade dedifferentiated liposarcomas. Int J Cancer 2002;99:68–73.
Mentzel T, Van Der Berg E, Molenaar WM . Myxofibrosarcoma. In: Fletcher CDM, Krishan Unni K, Mertens F (eds). World Health Organization Classification of Tumours. Pathology & Genetics of Tumours of Soft Tissue and Bone. International Agency for research on Cancer Press: Lyon, 2002, pp 102–103.
Mentzel T, Pedeutour F . Pleomorphic liposarcoma. In: Fletcher CDM, Krishan Unni K, Mertens F (eds). World Health Organization Classification of Tumours. Pathology & Genetics of Tumours of Soft Tissue and Bone. International Agency for Research on Cancer Press: Lyon, 2002, pp 44–45.
Kallioniemi A, Kallioniemi OP, Sudar D, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992;258:818–821.
Kallioniemi OP, Kallioniemi A, Piper J, et al. Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes Chromosomes Cancer 1994;10:231–243.
Chibon F, Mairal A, Freneaux P, et al. The RB1 gene is the target of chromosome 13 deletions in malignant fibrous histiocytoma. Cancer Res 2000;60:6339–6345.
Acknowledgements
This work was supported by grants from the INSERM and the Ligue Nationale Contre le Cancer (Comité de Paris).
Author information
Authors and Affiliations
Corresponding author
Additional information
Duality of interest
None declared.
Rights and permissions
About this article
Cite this article
Idbaih, A., Coindre, JM., Derré, J. et al. Myxoid malignant fibrous histiocytoma and pleomorphic liposarcoma share very similar genomic imbalances. Lab Invest 85, 176–181 (2005). https://doi.org/10.1038/labinvest.3700202
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700202
Keywords
This article is cited by
-
Systemic Therapy in Advanced Pleomorphic Liposarcoma: a Comprehensive Review
Current Treatment Options in Oncology (2023)
-
Establishment and characterization of NCC-PLPS2-C1: a novel cell line of pleomorphic liposarcoma
Human Cell (2022)
-
Prognostic value of CD34 expression status in patients with myxofibrosarcomas and undifferentiated pleomorphic sarcomas
Scientific Reports (2021)
-
Establishment and characterization of NCC-PLPS1-C1, a novel patient-derived cell line of pleomorphic liposarcoma
Human Cell (2021)
-
Sarcoma classification by DNA methylation profiling
Nature Communications (2021)
