
Abstract
Study design:
Case report of a patient with subacute delayed myelopathy after an acute low thoracic spine injury.
Objectives:
To draw awareness to a rarely described complication with potential to add devastating neurological insult to the original spinal cord injury, and to discuss evidence supporting a vascular mechanism.
Setting:
Health Science Centre, Winnipeg, Manitoba, Canada.
Case report:
A 35-year-old woman developed clinical and MRI evidence of ascending myelopathy, extending up to C5, 16 days after a T11/12 fracture dislocation. The distribution of MRI signal abnormality, MRI evidence of prominent venous markings, and association with upright mobilization and the wearing of a thoraco-lumbo-sacral orthosis, suggest that elevated spinal venous pressure in conjunction with low arterial pressure may have induced impaired spinal cord vascular perfusion.
Conclusion:
After recent spinal cord injury, factors exacerbating spinal venous hypertension and/or arterial hypotension may in some patients lead to impaired spinal cord perfusion. These factors should be considered and corrected if symptoms or signs of progressive myelopathy emerge in the first few days or weeks after injury.
Similar content being viewed by others


Introduction
Several types of neurological deterioration, not directly attributable to ongoing mechanical instability of the spine, may occur after acute spinal cord injury. During the first few hours and days, neurological deterioration may result from edema and other secondary changes involving one or two spinal cord segments adjacent to the original site of trauma. After a period of neurological stability, late delayed complications, such as progressive post-traumatic cystic myelopathy (syringomyelia)1, 2, 3 or progressive post-traumatic myelomalacic myelopathy,4, 5 may produce new ascending neurological symptoms months, years, or even decades after the original injury.
In addition to acute phase progression and late delayed syndromes, myelopathic deterioration may present during the first few weeks after spinal cord injury. Although several mechanisms have been postulated, the exact cause of subacute delayed post-traumatic myelopathy is unknown.
Case report
A 35-year-old woman suffered a hyperflexion injury of the lower thoracic spine resulting in complete loss of sensation and voluntary movement below T11. On admission, the cervical spine was cleared and she had no neck pain. Computerized axial tomography (CT) demonstrated a fracture-dislocation; the T11 vertebral body was displaced anteriorly by 50% on the T12 vertebral body, resulting in marked stenosis of the central canal and severe spinal cord compression (Figure 1). She was treated with intravenous methylprednisilone 30 mg/kg bolus followed by 5.4 mg/kg/h for 23 h. At 4 days after admission, she underwent open reduction and internal fixation from T9 to L1, without complication, and was started on low molecular weight heparin, 5000 units daily.

CT sagittal reconstruction of the spine showing a fracture dislocation. The T11 vertebral body is displaced anteriorly by approximately 50% on the T12 vertebral body
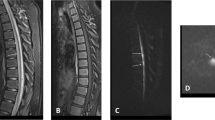
On postinjury day 14, she began to wear a thoraco-lumbo-sacral orthosis (TLSO) and sit at 45° for brief periods, during which time she experienced light-headedness. Her routine daily blood pressures, recorded in the supine position, ranged from 92/48 to 106/60 mmHg. During postinjury days 16 and 17, the level of numbness ascended, insidiously, to her mid chest. She also reported pain in the back, chest, and low-neck regions. Neurological examination confirmed that the sensory level had risen to the T5 level by postinjury day 17. A magnetic resonance imaging (MRI) study of the cervical and thoracic spine on postinjury day 18 showed increased T2-weighted signal from C5 to T9 (Figure 2a; artefact from spinal instrumentation obscured visualization of cord segments caudal to T9). The abnormal signal, and cord swelling, was most intense in the lower cervical and upper thoracic segments. Axial sections demonstrated that the signal abnormality was located in the posterior cord region (Figure 2b and c). There was no gadolinium enhancement. Prominent vascular markings were noted in the thecal sac in the thoracic region (Figure 3), suggesting elevated venous pressure. Thoracic and abdominal aortograms, as well as selective injections of the spinal arteries from T8 to L2 showed no evidence of thrombosis, vascular anomaly, or dural arteriovenous fistula to account for evidence of distended vascular markings noted on MRI. Plain radiographs of the thoracolumbar spine showed no change in the spine alignment or position of the fixation hardware. An MRI of the brain, chest radiograph, lung scan, abdominal CT and sonogram, and duplex venous sonogram of both legs were normal. Blood tests included a normal complete blood cell count, INR, PTT, sedimentation rate, C-reactive protein, antiphospholipid antibody screen, and antinuclear antibody screen. Pain subsided over several days without specific treatment. Repeat MRI of the cervical and thoracic spine on postinjury day 22 showed substantial decrease of the T2-weighted signal abnormality. By 7 months postinjury, MRI of the cervical cord was normal, however, marked atrophy of the thoracic cord caudal to T5 was noted; clinically a T5 sensory level persisted.

MRI of the spinal cord on postinjury day 18. (a) T2-weighted sequence shows increased signal and swelling in the cervical and upper thoracic spinal cord. (b) Axial T2-weighted image shows increased signal in the central and posterior cord region. (c) T1-weighted image shows decreased signal in the central and posterior cord region

MRI of the spinal cord on postinjury day 18 shows prominent vascular markings in the thecal sac
Discussion
Delayed neurological deterioration during the first month after lower spinal cord injury has been rarely reported. In 1969, Frankel described eight patients with post-traumatic ascending myelopathy starting 2–11 days after injury and progressing for 2–12 days.6 They all had fractures of the lower thoracic spine, except one patient with a T5 fracture. More recently, Belanger et al,7 reported ascending neurological deficit in three patients with spine injuries, starting 7–13 days after injury.7 Similar to the present patient, one of their cases had a low spinal injury (L1 level) and showed increased T2-weighted MRI signal in the central cord region extending to the mid cervical level. Also, very similar to the present patient, is the 30-year-old woman with a fracture dislocation at T11/12 described by Aito et al.8 Low baseline blood pressures were noted in their patient, and on postinjury day 12, she developed scapular pain and paresthesia in the upper limbs whenever she sat upright for physiotherapy. Two days later her sensory level ascended to the T3 level and an MRI showed increased T2-weighted signal between C3 and T1. Visocchi et al9 reported two patients with post-traumatic ‘descending’ myelopathy, which they attributed to spinal venous thrombosis in one patient and anterior spinal artery occlusion (secondary to disk herniation) in the other patient.
Unlike the distinctly rare occurrence of subacute delayed myelopathy after low spinal injury, early delayed deterioration is noted more frequently (6–10%) in patients with acute cervical spine trauma.10, 11 In the series by Harrop et al,10 approximately half of the patients who deteriorated developed new symptoms within the first 24 h of injury, in association with halo vest placement or cervical spine traction immobilization. Most of the other patients deteriorate 1–7 days after cervical trauma; the mechanism was unknown but the authors suggested the possibility of cord ischemia secondary to sustained hypotension and/or venous insufficiency.
To date, most patients with lower spine trauma and delayed ascending myelopathy, including our patient, were injured near the thoraco-lumbar junction. This part of the spine corresponds to the region most frequently entered by the Great Artery of Adamkiewicz (GAA), the largest single medullary artery supplying the spinal cord.12 Thus, post-traumatic thrombus of the GAA was considered by Aito et al,8 among other etiologies, as a possible cause of early delayed ascending myelopathy. However, a spinal angiogram was not performed on their patient. Angiographic study of the present patient, as well as the patient with an L1 injury reported by Belanger et al,7 showed no evidence of thrombus in the GAA.
Impaired venous drainage of the cord after injury may contribute to early delayed myelopathy.8, 10 After spinal cord injury, 68% of patients demonstrate reversal of the normal hemodynamic gradient which in healthy subjects causes blood to flow from the paravertebral venous plexus to the inferior vena cava.13 The gradient reversal is thought to be due to increased vena caval pressure which, in turn, produces retrograde elevation of pressure and decreased venous flow in the valveless paravertebral system.13 Although variable in distribution, venous stasis and infarction involves predominantly central and posterior cord regions in autopsied cases.14, 15 Lesions produced by experimental occlusion of the dorsal spinal vein of the rat16 and monkey17 are confined to the posterior columns. These observations, in conjunction with the central posterior cord distribution of MRI signal abnormality in the present patient and the presence of distended venous markings in the thecal sac, suggests that elevated venous pressures causing venous stasis and ischemia, may have contributed to the subacute delayed myelopathy.
However, if increased spinal venous pressure and stasis is relatively common among patients with spinal cord injury,13 why is early delayed myelopathy only rarely reported? Other factors must be important.
One consideration is the effect of mobilization after injury. The present patient, and the patient described by Aito et al,8 developed symptoms when they began to sit upright. Increased intraabdominal pressure associated with sitting, and wearing a TLSO, may further impair venous return from the paravertebral plexus. Sitting may also induce an orthostatic drop in blood pressure, including spinal arterial pressure, especially in patients with spinal cord injury. Compatible with this mechanism, Cybulski, and D'Angelo described four patients who underwent laminectomy for nontraumatic cervical spondylosis and postoperatively developed myelopathy in association with hypotensive episodes (50–60 mmHg systolic) evoked while attempting to sit.18
The combination of decreased spinal arterial pressure and increased spinal venous pressure, potentially aggravated by sitting and the use of a TLSO, may impair cord perfusion sufficient to produce an ischemic insult. Consistent with this hypothesis, a combination of decreased spinal arterial pressure (secondary to transient moderate hypotension) and elevated vertebral venous pressure (associated prone positioning and use of chest rolls) was previously proposed as the cause of transient cervical myelopathy in four patients who underwent elective laminectomy for myeloradiculopathy.19
In conclusion, the observations suggest that factors exacerbating spinal venous hypertension and/or arterial hypotension may in some instances lead to impaired spinal cord perfusion. These factors should be promptly considered and corrected if symptoms or signs of progressive myelopathy emerge during the first few days or weeks after spinal cord injury.
References
Quencer RM, Green BA, Eismont FJ . Post-traumatic spinal cord cysts: clinical features and characterization with metrizamide computed tomography. Radiology 1983; 146: 415–423.
Rossier AB et al. Posttraumatic cervical syringomyelia. Brain 1985; 108: 439–461.
Vernon JD, Silver JR, Ohry A . Post-traumatic syringomyelia. Paraplegia 1982; 20: 339–354.
Falcone S et al. Progressive posttraumatic myelomalacic myelopathy: Imaging and clinical features. Am J Neuroradiol 1994; 15: 747–754.
Gebarski SS et al. Posttraumatic progressive myelopathy. Radiology 1985; 157: 379–385.
Frankel HL . Ascending cord lesions in the early stages following spinal injury. Paraplegia 1969; 7: 111–118.
Belanger E et al. Subacute posttraumatic ascending myelopathy after spinal cord injury. J Neurosurgery 2000; 93: 294–299.
Aito S et al. Ascending myelopathy in the early stage of spinal cord injury. Spinal Cord 1999; 37: 617–623.
Visocchi M, Di Rocco F, Meglio M . Subacute clinical onset of posttraumatic myelopathy. Acta Neurochir 2003; 145: 799–804.
Harrop JS et al. The cause of neurological deterioration after acute cervical spinal cord injury. Spine 2001; 26: 340–346.
Yablon IG et al. Acute ascending myelopathy of the spine. Spine 1989; 14: 1084–1089.
Sliwa JA, Maclean IC . Ischemic myelopathy: a review of spinal vasculature and related clinical syndromes. Arch Phys Med Rehab 1992; 73: 365–372.
Cassar-Pullicino VN et al. Hemodynamic alterations in the paravertebral venous plexus after spinal injury. Radiology 1995; 197: 659–663.
Hughes JT . Venous infarction of the spinal cord. Neurology 1971; 21: 794–800.
Kim RC et al. Nonhemorrhagic venous infarction of the spinal cord. Ann Neurol 1984; 15: 379–385.
Zhang Z et al. Circulatory disturbance of rat spinal cord induced by occluding ligation of the dorsal spinal vein. Acta Neuropathol 2001; 102: 335–338.
Doppman JL, Girton M, Popovsky MA . Acute occlusion of the posterior spinal vein. J Neurosurg 1979; 51: 201–205.
Cybulski GR, D'Angelo CM . Neurological deterioration after laminectomy for spondylotic cervical myeloradiculopathy: the putative role of spinal cord ischaemia. J Neurol Neurosurg Psychiatry 1988; 51: 717–718.
Bhardwaj A et al. Neurological deficits after cervical laminectomy in the prone position. J Neurosurg Anesthesiol 2001; 13: 314–319.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Schmidt, B. Subacute delayed ascending myelopathy after low spine injury: case report and evidence of a vascular mechanism. Spinal Cord 44, 322–325 (2006). https://doi.org/10.1038/sj.sc.3101801
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.sc.3101801
Keywords
This article is cited by
-
Very rare incidence of ascending paralysis in a patient of traumatic spinal cord injury: a case report
Spinal Cord Series and Cases (2022)
-
A rare cause of neurological deterioration to complete paraplegia after surgery for thoracic myelopathy: a case report
Spinal Cord Series and Cases (2019)
-
Subacute posttraumatic ascending myelopathy: a literature review
Spinal Cord (2017)
-
Subacute post-traumatic ascending myelopathy after T12 burst fracture in a 32-year-old male: case report and surgical result of cervical durotomy
Spinal Cord Series and Cases (2016)
-
Subacute delayed ascending myelopathy: not just a post-traumatic disorder
Spinal Cord (2014)
