
Abstract
Increasing evidence has indicated that glycans and their interactions with glycan-specific binding proteins have crucial roles in most physiological processes. One emerging topic in biomaterials is the construction of smart materials capable of recognizing and responding to these valuable glycans. However, because of the complicated compositions and structures as well as the low abundances and microheterogeneity of glycans, developing glycan-responsive materials with a high sensitivity and specificity is a long-term, challenging goal. Here, we report a biomimetic polymer capable of capturing and discriminating sialo-complex-type glycans with the core Neu5Ac-Gal-GlcNAc-Man sequence, which is one of the most effective biomarkers for cancer detection. As an optimized dipeptide, L-Asp-L-Phe has a strong but differential binding affinity for N-acetyl-neuraminic acid (Neu5Ac), N-acetyl-glucosamine (GlcNAc), galactose (Gal), and mannose (Man), which are the core construction units of sialylated glycans. In addition, the L-Asp-L-Phe-grafted polyethyleneimine film displays remarkable adsorption behavior for the model sialylated glycans and can discriminate their linkage isomers, which is accompanied by significant changes in the surface morphology and stiffness. These features facilitate the highly selective capture of sialylated glycopeptides from complex protein samples using PEI-g-L-Asp-L-Phe-modified mSiO2@SiO2@Fe3O4 core-shell microspheres. The mechanism analysis demonstrates that the favorable polymeric spatial structures and multiple synergetic hydrogen-bonding interactions among the dipeptides and glycan are the main driving forces, indicating a clear direction for designing glycan-specific biomaterials.
Similar content being viewed by others
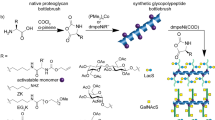
Introduction
As essential biological components, saccharides, oligo-/polysaccharides, glycoconjugates, glycolipids, glycopeptides, and glycoproteins participate in most physiological processes and have crucial roles in energy metabolism,1 enzyme reactions,2 immune response,3 cell communication and adhesion,4 fertilization,5 cell migration6 and the development of tissues and organs.7 In these physiological activities, glycans act as important bio-information carriers, immune recognition executors and cell function regulators. Slight alterations in the glycan composition or structure, such as the absence of a sialic acid residue or an increase in bis-GlcNAc branching, could result in remarkable changes in protein conformation and function.8 The most well-known example of this is the terminal tri-saccharides of the glycoprotein antigen located on the erythrocyte membrane that determine the human ABO blood types.9 In addition, aberrant protein glycosylations are closely associated with tumor metastasis,10 immunological diseases11 and neurological disorders.12 For example, the overexpression of α-2,3-sialyted biantennary N-linked glycans is found in patients with prostate cancers,13 and the α-2,6-sialyted glycans exist in breast cancer patients.14 Therefore, if these valuable glycans can be detected with an adequate sensitivity, they can be used as biomarkers in early diagnosis of cancer15 or tumor cell separation,16 as molecular targets in new drug development17 and for the fabrication of glycan-specific drug-release systems.18
However, glycan structures are highly complicated owing to their anomeric configuration, alkane dehydrocyclization and glycosylation heterogeneity that are expressed by specific building blocks, different linkage types and characteristic glycoform patterns.19 These features make glycan targets indistinguishable from their analogs. Meanwhile, in eukaryotes, glycans are usually found on the exterior surface of cells with an extremely low abundance, and the surrounding biomolecules (for example, proteins, liposomes and oligosaccharides) with a high abundance cause serious background interference with the detection of the targeted glycans.20 Considering the complexity of glycosylation, developing glycan-specific materials or devices is very difficult, and the development of materials for recognizing and responding to these glycans in human cytomembrane or intracellular microenvironments will be more challenging. Thus far, a large number of saccharide-responsive materials or surfaces have been reported, including those based on reversible boronic acid–diol complexation21 and pH-sensitive materials triggered by glucose oxidase.22 However, these classical systems mainly focus on one or two active monosaccharides located on the terminal of a glycan, and they usually ignore the complete characteristics of the glycan, which contain a wealth of biological information. In comparison, to the best of our knowledge, glycan-targeted smart polymers are seldom reported. Here, for the first time, a biomimetic design idea was proposed to develop smart polymers with a precise recognition capacity for sialo-complex N-linked glycan.
N-linked glycosylation is the most thoroughly studied form of glycosylation. All N-linked glycans, including high-mannose, hybrid and complex types, are based on the common core pentasaccharides Man3GlcNAc2.23 Scheme 1a displays the bi-antennary form of the complex-type glycan that contains terminal sialic acid residues and bisecting GlcNAc-β(1→4)Gal units at both the α-3 and α-6 mannose sites on the pentasaccharide core. This type of glycan is an effective biomarker for pancreatic cancer detection.24 With these characteristics in mind, the key to constructing a complex-type glycan-targeted material is designing a molecular receptor capable of recognizing and discriminating the core glycan fragment composed of four monosaccharide residues, namely, Neu5Ac-Gal-GlcNAc-Man. In this respect, nature has inspiration for the saccharide receptor design, for example, Maackia amurensis leukoagglutinin displays a strong but specific binding with sialylated tri-saccharide (that is, Neu5Ac-α(2-3)Gal-β(1-4)GlcNAc).25 Multiple, complementary hydrogen-bonding interactions are formed between the tri-saccharide and the specific amino-acid residues located at different positions on the protein (Scheme 1b). This feature inspires us to design an oligopeptide-based saccharide receptor and integrate it into a smart polymer system to mimic the intermolecular complexation between a lectin and saccharide.
Our previous study systematically investigated the discrimination capacities of 54 types of dipeptides for diverse monosaccharides, and we discovered that dipeptide sequences with a combination of hydrophobic and hydrophilic amino-acid residues had optimal performances for saccharide separation.26 Directed by this dipeptide-screening strategy, in this work, we introduced L-Asp-L-Phe as an optimized saccharide receptor. The hydrogen nuclear magnetic resonance (1H NMR) titration experiments indicated that this dipeptide has a strong and differential binding affinity for Neu5Ac, Gal, GlcNAc, Man or Glc. Then, L-Asp-L-Phe was grafted onto a branched polyethyleneimine (PEI) main chain (average molecular weight of 10 000 Da) with a grafting ratio of ~6% to construct a smart polymeric network,27 and more details of the synthetic process are displayed in Supplementary Scheme 1. A quartz crystal microbalance with a dissipation monitoring (QCM-D) experiment and corresponding surface stiffness analysis demonstrated that the prepared PEI-g-L-Asp-L-Phe (abbreviated PEI-g-DF) thin film displayed satisfactory chemoselectivity for three glucose homologs that only differ in the stereo-orientation of a hydroxyl as well as remarkable adsorption with Neu5Ac, Gal and GlcNAc, which further facilitated the highly selective adsorption of the sialyllactose linkage isomers28 (that is, core fragments of the sialo-complex-type glycans) and N-linked disialylated bi-antennary glycan (that is, the real glycan appended to a glycopeptide). Furthermore, PEI-g-DF was chemically immobilized on a mSiO2@SiO2@Fe3O4 core–shell microsphere surface29 (Scheme 1d). The excellent glycan discrimination, strong adsorption capacities, high specific surface area and magnetic characteristics of our material contributed to its excellent performance in glycopeptide capture,16 and a high proportion of sialo-complex-type glycopeptide was identified in tryptic digests of fetuin mixed with a 200-fold bovine serum albumin (BSA) interference, which validated our glycan-target design concept.
Materials and methods
Preparation of PEI-g-DF
The L-Asp-L-Phe (DF) was synthesized on Fmoc-L-Phe Wang resin (95%, GL Biochem., Shanghai, China) via a peptide synthesizer (CS 136XT, CS-Bio, CA, USA) utilizing standard solid-phase synthesis procedure. After being cleaved and side-chain-deprotected from resin, the crude product was purified through a high-performance liquid chromatography (UFLC-20 A purity system, Shimadzu, Kyoto, Japan) with a reversed-phase chromatographic column (Inertsil ODS-SP C18: 4.6 mm × 250 mm, 5 μm). Then, the purified DF was dispersed in the methanol solution with a few drops of dimethysulfoxide in ice bath and stirred for 2 h, obtaining the dimethyl-esterified DF for the following experiment. After chloride acetylation and subsequent purification by a silica gel column, the amidated dipeptide units were grafted onto the PEI (MW=10 000, 99%, Aladdin, Shanghai, China) through a substitution reaction. The final product was dialyzed (HT0934, Mwco3.5KD × MD34 mm, Yobios, Xi′an, China) against methanol to remove the unreacted reagents for obtaining the pure PEI-g-DF. The branched PEI was used to provide sufficient amine- or imine-anchoring sites for grafting the dipeptide units. More detailed information about the preparation of the PEI-g-DF and PEI-g-DF@mSiO2@SiO2@Fe3O4 particles is listed in the Supporting Information.
1H NMR titration experiment
An 1H NMR (Mercury VX 300-MHz, Varian, CA, USA) titration experiment was adopted as a label-free method to investigate the binding affinity of the dipeptide (DF) for diverse saccharides, that is, Neu5Ac, Gal, GlcNAc, Man and Glc (Alfa Aesar, Tianjin, China). In this experiment, D2O was not used as the solvent because of the side effect of the acid–alkane interaction and the absence of active hydrogen protons. Thus, deuterated dimethylsulfoxide (d6-DMSO) with a high polarity similar to that of D2O was chosen as the solvent for the 1H NMR measurement. The association constants (Ka) were obtained based on the relationship between the chemical shifts of the amide-a H-proton of the dipeptide and the molar ratios of the monosaccharides to DF using a nonlinear calculation equation (Supplementary equation (1)).
Saccharide-selective adsorption study
Saccharide sensors were fabricated by electrostatic adsorption of the PEI-g-DF polymer on the Au surfaces of the QCM resonators. The selective adsorption behaviors of PEI-g-DF for seven types of saccharides (50 mM) in water, including the dynamic adsorption and dissipation curves, were recorded by a quartz crystal microbalance with dissipation monitoring (QCM-D, Q-Sense E4 System, Biolin Scientific AB, Stockholm, Sweden). The corresponding adsorption mass change on the resonator was calculated by Sauerbrey’s equation (Supplementary equation (2)). As a control experiment, the adsorption of various saccharides on the dipeptide-based homopolymer thin film (Poly-DF) was investigated to illustrate the rationality of the smart polymer design (Supplementary Figure 3). In addition, the adsorption behaviors of the pure PEI thin film interacting with monosaccharides were recorded as a control experiment, as shown in Supplementary Figure 4.
Surface morphology and stiffness characterization
Here, a PEI-g-DF-modified QCM-D resonator was used directly for investigating the surface morphology and stiffness change in the polymer thin film in response to the adsorption of monosaccharides or 3′-sialyllacotose. The corresponding changes in the surface morphology and stiffness were monitored using atomic force microscopy (Multimode 8 AFM, Bruker, MA, USA) in the peak force quantitative nano-mechanical mode. The PEI-g-DF-modified QCM-D resonators before and after interactions with different saccharides are displayed in the manuscript. In addition, the surface morphology of the pure PEI polymer thin film before and after interaction with 3′-sialyllacotose is displayed in Supplementary Figure 5 as the control experiment.
Bio-attenuated total reflectance Fourier transform infrared spectroscopy spectral characterization
Infrared spectra were recorded on a Fourier transform infrared spectroscopy (FT-IR) spectrometer (Vertex 80v, Bruker, MA, USA) in combination with a bio-attenuated total reflectance (Bio-ATR) cell II accessory. PEI-g-DF (240 mg ml−1) and 3′-sialyllactose (100 mg ml−1) were dissolved in deuterated dimethylsulfoxide (d6-DMSO). Before each measurement, the equipment equilibrated for 15 min to ensure the temperature was 25 °C, and then, the spectra of the samples (equivalent volume mixture of PEI-g-DF with 3′-sialyllactose) were obtained by 1200 scans and subtracting the d6-DMSO background at a 4 cm−1 resolution. All the 3′-sialyllactose and 6′-sialyllactose used in the above experiments were sodium salts. To obtain a clear observation of the stretching vibration of the hydroxyl groups (range from 3500 to 3000 cm−1), d6-DMSO was chosen as an alternative to D2O.
Results and Discussion
First, a 1H NMR titration experiment was performed to evaluate the binding affinity of the dimethyl-esterified L-Asp-L-Phe with Neu5Ac, Gal, GlcNAc and Man, which are the essential construction units of sialo-complex-type glycan. To obtain the detailed chemical shift changes of the active hydrogen protons (for example, amide, amine or hydroxyls), d6-DMSO was chosen as the solvent instead of D2O because of its high solvent polarity, which is comparable to that of D2O, and the good solubility of both the dipeptide and saccharide in it. Figure 1a shows the 1H NMR spectra changes of the dipeptide upon additions of different molar amounts (0~10 equiv.) of Neu5Ac in d6-DMSO. A clear upfield shift from 8.98 to 8.92 p.p.m. was observed for the amide-a H-proton of the dipeptide, which can be attributed to a hydrogen-bonding interaction,30 whereas the terminal amine-b H-proton signal of the dipeptide gradually became weaker and finally disappeared when 1 equiv. of Neu5Ac was added, which might correspond to an acid–base reaction between the carboxyl group in Neu5Ac and the amine in the dipeptide.31 It is worth noting that the 1H NMR shift of the amide-H proton might be an accumulative effect of the complicated binding modes32 between the dipeptide and Neu5Ac, which feature multiple hydroxyl groups capable of functioning as both hydrogen-bond donors and acceptors.33 According to a nonlinear fitting calculation34 (red line in Figure 1b) for the changes in the chemical shifts of the dipeptide based on the host-to-guest molar ratio, an association constant (Ka) of 595 M−1 was obtained for the dipeptide interacting with Neu5Ac. Their possible binding mode was described by a quantum chemical calculation (Gaussian, density function theory at the 6-311G level, solvent: water, Figure 1f), which was well consistent with the 1H NMR data. With the same method, the Ka value of the dipeptide interacting with Gal or GlcNAc (two neutral monosaccharides) was determined to be 275 or 195 M−1, respectively. The satisfactory Ka values and the chemical shift changes of the amide H-protons (Figures 1c and d) revealed that hydrogen-bonding interactions are the main driving force for complexation. In addition, L-Asp-L-Phe displayed a good chemoselectivity toward Neu5Ac, Gal, GlcNAc, Man and Glc (Figure 1e). Particularly, the difference in the interactions with Gal, Man and Glc was notable because these hexoses only differ in the stereo-orientation of one hydroxyl group, which is difficult to chemically recognize.

Binding affinity evaluation of L-aspartate-L-phenylalanine dimethyl ester (abbreviated DF) complexed with five core monosaccharides. (a) Partial hydrogen nuclear magnetic resonance (1H NMR) spectra from the titration of DF (60 mM) with different molar ratios (0~10) of Neu5Ac in deuterated dimethylsulfoxide (d6-DMSO) at 25 °C. (b–d) Relationship between the chemical shift of the amide-a proton and the molar ratios of the monosaccharide (guest) to DF obtained from the hydrogen nuclear magnetic resonance (1H NMR) titration experiment. Guests: Neu5Ac (b), Gal (c) and GlcNAc (d). The red lines are nonlinear fitting curves, which were used to calculate the association constants (Ka). (e) Comparison of Ka for DF interacted with Neu5Ac, Gal, GlcNAc, Man or Glc, illustrating the satisfactory chemoselectivity of the DF receptor for different monosaccharides. (f) Possible binding mode of DF with Neu5Ac obtained from quantum chemistry calculations (Gaussian, density function theory (DFT) at the 6-311G level, solvent: water); hydrogen bonds with different lengths are indicated by the green dashed lines, which are well consistent with the chemical shift changes in the 1H NMR spectra (b).
Although the binding affinity between a dipeptide and a monosaccharide is weak, this force increases remarkably when a functional polymer with sufficient binding sites and a favorable conformation is introduced. Therefore, the L-Asp-L-Phe units were grafted onto a branched PEI (average molecular weight: 10 000) through a substitution reaction, and the grafting density of ~6% was determined using the 1H NMR spectrum (Supplementary Figure 1) and an elemental analysis. Through electrostatic adsorption for 3 h, the prepared PEI-g-DF was anchored onto a gold layer of a QCM-D resonator with an average film thickness of 7 nm (Supplementary Figure 2), which remained stable during the following experiments. Then, a QCM-D adsorption experiment, which is a reliable surface-sensitive technology capable of providing real-time information about variations in the mass and viscoelastic properties of a layer,35 was performed to monitor the dynamic adsorption processes of diverse mono- and tri-saccharides on the PEI-g-DF surface. As shown in Figure 2a, the distinct frequency variation (ΔF) curves corresponding to the adsorption of Gal, Man and Glc indicated that this polymer film displayed a satisfactory chemoselectivity toward these glucose homologs. At 3 h, the adsorption capacity of Gal (Δm=601.2 ng cm−2) was substantially larger than that of Man (Δm=123.8 ng cm−2) or Glc (Δm=58.9 ng cm−2) on the polymer surface. Notably, the order of the monosaccharide adsorption capacity was in agreement with the results from 1H NMR, which revealed that the precise discrimination capacity of the polymer film originated from the dipeptide unit.36

Dynamic adsorption curves of various monosaccharides or sialo-trisaccharides on a polyethyleneimine-grafted-L-aspartate-L-phenylalanine dimethyl ester (PEI-g-DF) thin film monitored by a quartz crystal microbalance with dissipation (QCM-D). (a–d) Comparison of the time-dependent QCM frequency changes of the saccharides adsorbed on the PEI-g-DF-modified QCM resonators; (a) glucose homologs (three important biological hexoses), namely, Glc, Man and Gal; (b) core monosaccharide building blocks of complex-type N-linked glycan: Neu5Ac, GlcNAc and Gal; (c) Neu5Ac and its sodium salt; (d) glycosidic linkage isomers of sialyllactose, and their chemical structures are shown in f. (e) Comparison of the time-dependent dissipation changes of 3′- and 6′-sialyllactose on the PEI-g-DF-modified resonators. Saccharide concentrations: 50 mM; solvent: pure water, flow rate: 0.200 ml min−1; temperature: 25 °C.
Then, the dynamic adsorption behaviors of the core Gal, GlcNAc and Neu5Ac were investigated. The PEI-g-DF film displayed strong but differential adsorptions for these monosaccharides (Figure 2b). In addition to Gal, which had a ΔFmax of −102 Hz, the ΔFmax induced by GlcNAc or Neu5Ac adsorption reached −58 Hz (Δm=341.9 ng cm−2) or −41 Hz (Δm=241.6 ng cm−2), respectively. All of these values are substantially larger than those for adsorption on dipeptide-based homopolymers37 (less than 10 Hz; Supplementary Figure 3). This revealed the remarkable advantage of the smart polymer design. Neu5Ac shows a rapid adsorption process, and equilibrium was reached after 30 min, which can be attributed to electric adsorption driven by the complexation between the carboxyl group of Neu5Ac and the abundant amines of PEI. In comparison, hydrogen-bonding interactions also have important roles in Neu5Ac adsorption, as illustrated in Figure 2c. When the Neu5Ac anion (with sodium as the cation) was introduced at the same concentration to perform the control experiment, a transient sharp decrease (ΔF: −10 Hz) was observed in the first 10 min, and the frequency gradually decreased and reached −32 Hz after 3.5 h. In addition, a control experiment using pure PEI as the polymer layer was conducted, and the monosaccharide-induced adsorption was slight (ΔFmax ranging from −10 to −20 Hz) and lacked any evident differences (Supplementary Figure 4). This demonstrated that PEI only supplies a flexible polymer network and sufficient anchoring sites for the dipeptide recognition units.
Then, the research was expanded from basic monosaccharides to complicated, sialylated tri-saccharides, namely, Neu5Ac-α(2→3)-Gal-β(1→4)-Glc sodium salt (also called 3′-sialyllactose) and Neu5Ac-α(2→6)-Gal-β(1→4)-Glc sodium salt (6′-sialyllactose). Their chemical structures are shown in Figure 2f, and they can be regarded as the terminal fragments of the sialo-complex-type glycan. Figure 2d displays the dynamic adsorption curves of 3′- and 6′-sialyllactose on the PEI-g-DF thin film. Electrostatic adsorption and hydrogen-bonding interactions simultaneously dominated the initial adsorption stage (0–15 min), which resulted in rapid decreases in the QCM frequency for both 3′ and 6′-sialyllactose. After that, different chemical adsorption-dominated frequency changes were observed. For 3′-sialyllactose, the frequency changed from −34 to −52 Hz after 200 min. However, the frequency only changed from −15 to −20 Hz when 6′-sialyllactose was monitored. This indicated that PEI-g-DF had a precise discrimination capacity for the glycosidic-linkage isomers of the sialylated glycans. This result is noteworthy because this pair of glycan linkage isomers has been shown to have distinct roles in many pathological processes, but they are difficult to chemically discriminate. Meanwhile, as an important parameter reflecting the conformation, thickness and viscoelasticity of the polymer film, the dissipation curves for this pair of sialylated tri-saccharides were simultaneously recorded. As shown in Figure 2e, a larger dissipation change (ΔD) demonstrated that the polymer chains stretched into more relaxed and swollen states when the polymer film interacted with 3′-sialyllactose (ΔD: 14.4 × 10−6) than they did with the 6′-linkage isomer (ΔD: 6.8 × 10−6).
To verify the conformation change of the polymer chains, the corresponding morphological and mechanical property changes of the polymer film were investigated by AFM in a peak force quantitative nano-mechanical mode38 using an SNL-10 A tip (spring constant: 0.35 N m−1). In this experiment, the PEI-g-DF-adsorbed QCM-D resonator was used directly. Figure 3a shows the initial morphology of the polymer film, which was rather smooth with an average fluctuation of less than 2 nm, and numerous tiny particles attributable to polymeric aggregates were observed in the magnified image. After treatment with a Neu5Ac sodium salt aqueous solution (concentration: 50 mM) for 1 h and a subsequent washing process with water to remove the unbound Neu5Ac residues, a large number of small, polymeric bumps were observed in the large-scale AFM image, resulting in a substantial increase in the film fluctuations from 2.0±0.3 to 6.5±0.5 nm (Figure 3b). A similar phenomenon was also detected when the polymer film was treated with a Gal solution (Figure 3c). In comparison with the above monosaccharides, 3′-sialyllactose promoted more remarkable morphological changes, and several large bumps with an average fluctuation of 9±1 nm were observed clearly (Figure 3d), which can be attributed to the remarkable swelling behavior of the polymeric chains. Furthermore, the corresponding surface stiffness changes in the polymer film before (Figure 3e) and after being immersed in the 3′-sialyllactose solution (Figure 3f) were recorded using Young’s modulus maps. The remarkable color change from purple (corresponding to a high modulus) to yellow (a low modulus) indicated that the polymer surface became substantially softer after interacting with 3′-sialyllactose. This was further proven by the statistical analysis of the distribution of Young’s moduli according to the full images (insets in Figures 3e and f). The mean Young’s modulus of the polymer film decreased from 210 MPa to ~138 MPa. In comparison, there was no change in the surface morphology when the PEI thin film interacted with 3′-sialyllactose (Supplementary Figure 5). These data were consistent with the QCM-D dissipation changes and clearly indicated that the swelling behaviors of the polymer film respond to the sialylated tri-saccharide adsorption.

Dynamic adsorption curves of the various monosaccharides or sialo-trisaccharides on a polyethyleneimine-grafted-L-aspartate-L-phenylalanine dimethyl ester (PEI-g-DF) thin film monitored by a quartz crystal microbalance with dissipation. Surface morphology and stiffness changes of PEI-g-DF. (a–d) Surface morphologies of the PEI-g-DF thin film before (a) and after treatment with Neu5Ac sodium salt (b), Gal (c) or 3′-sialyllactose sodium salt (d) solutions with a concentration of 50 mM at 25 °C detected by an AFM in a peak force quantitative nano-mechanical (PFQNM) mode. The section profiles along the green lines in the AFM images are shown in the bottom layers. (e, f) Young’s modulus images of the polymer film before (e) and after treatment with a 3′-sialyllactose sodium salt solution (f) as well as the distribution of the Young’s moduli corresponding to the images. Scale bar: 1 μm.
Furthermore, an N-linked, di-sialylated, bi-antennary glycan, consisting of 11 monosaccharides and identical to glycan appended to a glycopeptide (Figure 4b), was introduced to perform the QCM-D adsorption and AFM morphological experiments. Although only one-tenth of the 3′-sialyllactose amount (that is, 5 mM) was used in the adsorption experiment, this bi-antennary glycan still induced a remarkable QCM frequency change, that is, up to 28 Hz, as shown in Figure 4a. Obvious spots with an average film fluctuation of 3±1 nm (Figure 4c) further demonstrated that the polymer chains experienced swelling after interacting with the bi-antennary glycan.

N-linked, disialylated, bi-antennary glycan adsorption experiment on the polyethyleneimine-grafted-L-aspartate-L-phenylalanine dimethyl ester (PEI-g-DF) polymeric thin film. (a) Time-dependent QCM frequency change of the bi-antennary glycan adsorbed on the PEI-g-DF-modified resonators. Glycan concentration: 5 mM; solvent: pure water, flow rate: 0.200 ml min−1; temperature: 25 °C. (b) The chemical structure of the N-linked, disialylated, bi-antennary glycan. (c) The surface morphology of the PEI-g-DF film after treatment with the bi-antennary glycan. The section profile along the green line in the AFM image is shown in the bottom layer. Scale bar: 1 μm.
The above data demonstrated the strong adsorption capacity and satisfactory chemoselectivity of our polymer film for several monosaccharides, the core sialylated glycan fragments and the real bi-antennary glycan. A more challenging topic in glycobiology is how to selectively capture39 trace amounts of glycopeptides from highly complicated biosamples. To tackle this challenge, the prepared PEI-g-DF was chemically immobilized onto the surface of mSiO2@SiO2@Fe3O4 microspheres using its high surface specific area40 (204.8 m2 g−1) and magnetic features. In this system, the Fe3O4 nanoparticles function as the cores, and they were wrapped with a thin nonporous SiO2 layer to prevent Fe3O4 oxidation and nonspecific interactions with proteins.41 Then, a mesoporous SiO2 layer with a pore size of 1.8 nm was coated on the prepared SiO2@Fe3O4 surface and acted as a shell to improve the surface specific area. The pore size of 1.8 nm was chosen according to the literature, which noted that 2 nm was an appropriate pore size for peptide enrichment.42 More detailed characterization data are shown in Supplementary Figures 6–9. After grafting a monolayer of 3-(triethoxylsilyl)propyl isothiocyanate on the mSiO2@SiO2@Fe3O4 surface and the subsequent coupling reaction with the amine groups of PEI, the PEI-g-DF was immobilized onto the core–shell microsphere surface. The appearance of a high abundance of nitrogen (N) element signals and the increase in the oxygen (O) content in the X-ray photoelectron spectroscopy (Figure 5a), the inherited characteristic peaks (νC=O: 1659 cm−1 and δN–H: 1547 cm−1 in IR (Figure 5b)), a 14% weight loss in the thermal gravimetric analysis (Figure 5c), the increased mean hydrodynamic radius (Figure 5d), and the clear morphology and elemental distribution profiles obtained by scanning electron microscopy equipped with energy dispersive X-ray spectroscopy (Figure 5e) demonstrated that the PEI-g-DF was successfully immobilized onto the mSiO2@SiO2@Fe3O4 surface with a high grafting density.

Characterization data of the polyethyleneimine-grafted-L-aspartate-L-phenylalanine dimethyl ester (PEI-g-DF)-modified mSiO2@SiO2@Fe3O4 core–shell microspheres. (a–c) X-ray photoelectron spectroscopy (a), FT-IR spectroscopy (b) and thermal gravimetric analysis (c) of mSiO2@SiO2@Fe3O4 before (black lines) and after PEI-g-DF modification (red lines). (d) Comparison of the hydrodynamic radius distribution of Fe3O4, SiO2@Fe3O4, mSiO2@SiO2@Fe3O4 and PEI-g-DF@mSiO2@SiO2@Fe3O4 measured by dynamic light scattering in pure water. (e) Microscopic morphology and elemental distribution of PEI-g-DF@mSiO2@SiO2@Fe3O4 observed by a scanning electron microscope with energy-dispersive X-ray spectroscopy. The EDX spectra of mSiO2@SiO2@Fe3O4 before and after modification with PEI-g-DF are displayed in Supplementary Figure 10.
Then, the enrichment efficiency of the PEI-g-DF-grafted microspheres for glycopeptides was evaluated using tryptic digests of bovine fetuin (a standard glycoprotein) mixed with different levels of BSA (a typical non-modified protein). Before the enrichment, the whole mass spectrum (MS) was occupied by high-abundance, non-modified peptide signals, which resulted in no detectable glycopeptide signal. Through a stepwise elution procedure (Figure 6a) with the assistance of a magnetic field, the captured glycopeptides were easily separated from the peptide mixture using our functional magnetic material, which facilitated the efficient removal of most of the non-glycopeptides with 80% acetonitrile/50 mM ammonium formate (NH4FA, pH=3.8) as the eluent. When the solvent polarity was improved to 70% acetonitrile/50 mM NH4FA, the bound glycopeptides dissociated from the material surfaces and were detected by MS. Figure 6c displays the MS of the fetuin tryptic digests mixed with 10-fold BSA digest interference after enrichment by our material. After comparing the results with the glycopeptide database and analyzing them by tandem MS/MS,43 30 glycopeptide signals were identified, and among them, 23 glycopeptides possessed the typical structures of sialo-complex-type N-linked glycans. The other seven glycopeptides were equipped with a common pentasaccharide core. These results demonstrated that our material exhibited a high selectivity for the specific glycan with Neu5Ac-Gal-GlcNAc-Man units. Detailed information about the glycopeptide sequences and glycan compositions is shown in the Supplementary Table 1. Furthermore, the high selectivity of our material was further verified by capturing 14 sialylated glycopeptides from a tryptic digest mixture of fetuin with a 200-fold BSA interference, as shown in Supplementary Figure 11 and Supplementary Table 2. In addition, as a control experiment to validate the role of the dipeptide unit, a glycopeptide enrichment with PEI@mSiO2@SiO2@Fe3O4 microspheres of the tryptic digests of fetuin and BSA at a molar ratio of 1:10 was conducted, as shown in Supplementary Figure 12. Although 10 glycopeptides were identified by MS, the signal intensities were low. The MS spectrum was occupied by a large number of non-modified peptide signals with high intensities. This indicated that both the non-modified peptides and glycopeptides had strong interactions with the PEI@mSiO2@SiO2@Fe3O4, which resulted in its poor enrichment selectivity for sialylated glycopeptides. Therefore, it is reasonable to presume that the dipeptides appended in the PEI chains have indispensable roles in the sialylated glycan recognition and precise capture.

Glycopeptide enrichment by PEI-g-DF@mSiO2@SiO2@Fe3O4 microspheres. (a) Schematic illustration of the enrichment strategy based on a dispersive solid-phase extraction (dSPE) mode. (b) Possible binding mode of a typical bi-antennary sialo-glycopeptide on the polyethyleneimine-grafted-L-aspartate-L-phenylalanine dimethyl ester (PEI-g-DF) polymer surface (blue layer); hydrogen bonds are indicated by red dashed lines. (c) Mass spectra of glycopeptides enriched with our material from tryptic digests of fetuin and bovine serum albumin (BSA) at a molar ratio of 1:10. Detected glycopeptides are marked with their glycan structures: blue ▪: GlcNAc; green ●: mannose; yellow ●: galactose; red ▴: fucose; purple ♦: Neu5Ac. These glycopeptides were identified based on the ‘UniProt’ (peptide sequence) and ‘GlycoMod’ (glycan structures) databases. The corresponding serial numbers of the mass spectrum (MS) signals are labeled in the square brackets. A high proportion of the sialo-N-linked glycopeptides with core glycan fragment were identified, which validated our glycan-targeted design. More detailed information, including the m/z values of the identified glycopeptides and peptide sequences, is displayed in Supplementary Table 1.
In addition to the high enrichment selectivity, the smart polymer-based material displayed a high adsorption capacity for a standard sialylated glycopeptide, and the adsorption capacity of PEI-g-DF@mSiO2@SiO2@Fe3O4 was 240 mg g−1, which was substantially larger than that of PEI@mSiO2@SiO2@Fe3O4 (80 mg g−1), as shown in Supplementary Figure 13.
On the basis of the above results, a molecular mechanism was further explored to explain the intermolecular complexation between PEI-g-DF and the sialo-complex-type N-linked glycans. In the following experiments, a commercially available 3′-sialyllactose sodium salt was chosen to represent the characteristic sialylated glycan fragment. First, an isothermal titration microcalorimetry measurement was performed to evaluate the binding affinity of the sialylated glycan with our polymer. As shown in Figure 7a, an abrupt slope was observed in the isothermal titration microcalorimetry titration curve at the beginning of the 20 min titration of 3′-sialyllactose sodium salt (3 mg ml−1) into PEI-g-DF (12 mg ml−1), which indicated the complexation between PEI-g-DF and 3′-sialyllactose. With the continued addition of sialylated trisaccharides into the polymer solution, a second isothermal titration microcalorimetry transition appeared, which revealed a more complex formation driven by strong, multiple-site bindings. In addition, the association constants of Ka1=160±23 M−1, Ka2=704±72 M−1, Ka3=861±99 M−1 and Ka4=34±3 M−1 were obtained from a nonlinear fitting calculation of the isothermal titration microcalorimetry titration curve (Figure 7b), which resulted in a cumulative Ka (Ka1 × Ka2 × Ka3 × Ka4, ~3.3 × 109 M−1) representing the intensive complexation between 3′-sialyllactose and the polymer. Then, the binding details were investigated by a 1H NMR titration, and d6-DMSO was chosen as the solvent to better observe the chemical shift changes of the active hydroxyl protons of the 3′-sialyllactose sodium salt. As shown in Figures 7c–e, for an equivalent volume mixture of PEI-g-DF (50 mg mL−1) with 3′-sialyllactose (20 mg ml−1), most of the hydrogen protons of 3′-sialyllactose had chemical shift changes, such as a clear upfield shift from 8.26 to 8.18 p.p.m. for the amide proton (H1), a remarkable decrease in the intensity of the C–H proton signal (H28), a downfield shift from 6.11 to 6.16 p.p.m. for the hydroxyl proton (H4), and a combination of C–H protons (H6, H7) forming a broad peak, for the Neu5Ac residue. In addition, the hydroxyl protons (H16, H22, H25 and H27) in Gal and Glc also displayed slight upfield shifts and remarkable peak shape changes. From the perspective of PEI-g-DF, the amide proton signals of the dipeptide-binding units weakened, which further proved that multiple hydrogen-bonding interactions were the main driving forces for the strong complexation of the polymer with the glycan. This presumption was further validated through Bio-ATR-FT-IR in DMSO at 25 °C (Figures 7f and g). Before the interaction with PEI-g-DF, 3′-sailyllactose displays a broad peak centered at 3350 cm−1, which is attributed to the stretching vibration of the hydroxyl groups in the saccharide. After the interaction with the polymer, this broad peak became sharp and was accompanied by a clear redshift to 3416 cm−1 for the maximum adsorption. More remarkable changes were observed for the stretching vibration (ν) bands of the carbonyl groups (Figure 7g). The characteristic ν(C=O) peak of PEI-g-DF shifted from 1669 to 1665 cm−1, whereas the ν(C=O) peak of 3′-sialyllactose shifted from 1642 to 1649 cm−1. In addition, the δ(CONH) peak of the terminal Neu5Ac residue in 3′-sialyllactose displayed a slight shift from 1617 to 1615 cm−1. All these data indicated that the carbonyl, amide and hydroxyl groups in 3′-sialyllactose and the dipeptide carbonyl groups in the polymer participated in the complexation.

Binding affinity study of the 3′-sialyllactose sodium salt with polyethyleneimine-grafted-L-aspartate-L-phenylalanine dimethyl ester (PEI-g-DF). (a, b) Isothermal titration microcalorimetry data and fitting curve of 3′-sialyllactose (3 mg ml−1) titrated into PEI-g-DF (12 mg ml−1) in H2O at 25 °C. (c–e) Partial hydrogen nuclear magnetic resonance (1H NMR) spectra of PEI-g-DF (50 mg ml−1; c), 3′-sialyllactose (20 mg ml−1; d) and a mixture of PEI-g-DF with an equivalent amount of 3′-sialyllactose (e) in deuterated dimethylsulfoxide (d6-DMSO) at 25 °C. The chemical shift attribution for each hydrogen proton in 3′-sialyllactose is marked by a numerical symbol in its chemical structure. The chemical shift changes of the hydrogen protons are indicated by dashed black arrows or box. (f, g) Representative bio-attenuated total reflectance Fourier transform infrared spectroscopy (Bio-ATR-FT-IR) spectra of PEI-g-DF (black), 3′-sialyllactose (red) and its mixture with an equivalent amount of PEI-g-DF (blue) in d6-DMSO at 25 °C. Characteristic IR peaks and corresponding vibration attributions are indicated by black arrows. All the 3′-sialyllactose used in these experiments were a sodium salt to avoid the side effect of acid–alkaline interactions.
On the basis of the above 3′-sialyllactose-binding affinity analysis and its adsorption dynamic study, a possible glycopeptide-binding mechanism was proposed, as illustrated in Figure 6b. PEI-g-DF was grafted onto the surface of mesoporous SiO2 to construct a thin polymeric layer. When a sialylated glycopeptide approached the material surface, the exposed glycan fragments (that is, Neu5Ac-Gal-GlcNAc) can be captured by multiple dipeptide units grafted on the PEI main chains. Specifically, Neu5Ac, Gal and GlcNAc in the terminal end of glycan prefer to combine with the Asp-Phe-binding units through multiple hydrogen-bonding interactions between the hydroxyl or amide groups in the saccharide residues and the carbonyl or amide groups in the dipeptides, as indicated by the red dashed lines. The secondary amines of PEI might also participate in the binding with the glycan; however, PEI was presumed to have a more important role in constructing a favorable polymeric conformation, which allowed the grafted dipeptide units to better combine with the glycan.
Conclusions
Owing to the unique composition and structure characteristics of glycans that contain a wealth of bio-information and specific functions, developing glycan-targeted polymers can remarkably improve the accuracy and specificity of biomaterials to satisfy the demands raised by the ‘Precision Medicine Project’.44 In this study, using a sialo-complex-type N-linked glycan as a typical example, we reported a biomimetic design idea to construct a sialylated glycan-responsive polymeric material and successfully realized glycan-selective capture from a complicated peptide mixture sample. In contrast to the conventional design of saccharide-responsive materials that usually concentrates on one or two terminals or core monosaccharides, our study merged the glycan composition, structure and glycosidic linkage-type information into a dipeptide-based smart polymer design, which indicates a clear direction for the development of glycan-targeted biomaterials.45 It is worth mentioning that this study is just the beginning of new chapter, and more challenges need to be solved. In addition to the sialo-complex-type glycans reported here, the development of N-linked high mannose-type or hybrid-type glycans as well as O-linked glycan-targeted materials is substantially more difficult because it requires exquisite oligopeptide-sequence design and rational construction of a smart polymer system. In addition, developing glycan-sensitive materials capable of recognizing and responding to the addition or absence of a single monosaccharide in a glycan, such as those of blood-type-sensitive materials taking individual differences into account, is highly desirable and will help scientists better understand the mysterious world of glycans. Furthermore, emerging mesoporous materials, such as metal organic frameworks46 and covalent organic frameworks,47 integrated with smart polymer designs could remarkably increase the specificity and separation efficiency of the composite materials for the target glycans, which would facilitate a variety of exciting applications in the fields of bio-sensing and bio-separation.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.

Design idea for the glycan-specific responsive polymer. (a) Chemical structure of a typical sialo-complex-type N-linked glycopeptide; two Neu5Acα(2-3)Galβ(1-4) GlcNAc units are linked to a pentasaccharide core composed of Man3GlcNAc2. Precise recognition of the core sialo-saccharide fragment is key for the development of a sialylated glycan-specific receptor. (b) Unique binding model of Maackia amurensis lectin (PDB No: 1DBN) complexed with the sialo-oligosaccharide (Neu5Ac-α(2-3)Gal-β(1-4) GlcNAc); reproduced with permission.25 Copyright 2000, American Society for Biochemistry and Molecular Biology. Multiple hydrogen-bond interactions among the six amino-acid residues and guest oligosaccharide contribute to the high specificity, which inspired us to develop oligopeptide-based glycan receptors. (c) Chemical structure of a polyethyleneimine-grafted-L-aspartate-L-phenylalanine dimethyl ester (abbreviated PEI-g-DF). (d) Graphic illustration of the PEI-g-DF-modified mesoporous SiO2@SiO2@Fe3O4 core–shell microsphere.
References
Tremaroli, V. & Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249 (2012).
Koeller, K. M. & Wong, C.-H. Enzymes for chemical synthesis. Nature 409, 232–240 (2001).
Crocker, P. R., Paulson, J. C. & Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 7, 255–266 (2007).
Zhao, Y.-Y., Takahashi, M., Gu, J.-G., Miyoshi, E., Matsumoto, A., Kitazume, S. & Taniguchi, N. Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 99, 1304–1310 (2008).
Tulsiani, D. R. P., Yoshida-Komiya, H. & Araki, Y. Mammalian fertilization: a carbohydrate-mediated event. Biol. Reprod. 57, 487–494 (1997).
Guo, H.-B., Lee, I., Kamar, M., Akiyama, S. K. & Pierce, M. Aberrant N-glycosylation of β1 integrin causes reduced α5β1 integrin clustering and stimulated cell migration. Cancer Res. 62, 6837–6845 (2002).
Lastdrager, J., Hanson, J. & Smeekens, S. Sugar signals and the control of plant growth and development. J. Exp. Bot. 65, 799–807 (2014).
Song, Y., Aglipay, J. A., Bernstein, J. D., Goswami, S. & Stanley, P. The bisecting GlcNAc on N-glycans inhibits growth factor signaling and retards mammary tumor progression. Cancer Res. 70, 3361–3371 (2010).
Landsteiner, K. & Miller, C. P. Serological studies on the blood of the primates. J. Exp. Med. 42, 853–862 (1925).
Stowell, S. R., Ju, T. & Cummings, R. D. Protein glycosylation in cancer. Annu. Rev. Pathol. 10, 473–510 (2015).
Macauley, M. S., Crocker, P. R. & Paulson, J. C. Siglec regulation of immune cell function in disease. Nat. Rev. Immunol. 14, 653–666 (2014).
Freeze, H. H., Eklund, E. A., Ng, B. G. & Patterson, M. C. Neurological aspects of human glycosylation disorders. Annu. Rev. Neurosci. 38, 105–125 (2015).
Saldova, R., Fan, Y., Fitzpatrick, J. M., Watson, R. W. G. & Rudd, P. M. Core fucosylation and α 2-3 sialylation in serum N-glycome is significantly increased in prostate cancer comparing to benign prostate hyperplasia. Glycobiology 21, 195–205 (2011).
Lin, S., Kemmner, W., Grigull, S. & Schlag, P. M. Cell surface α2,6-sialylation affects adhesion of breast carcinoma cells. Exp. Cell Res. 276, 101–110 (2002).
Pinho, S. S. & Reis, C. A. Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15, 540–555 (2015).
Huang, C., Yang, G., Ha, Q., Meng, J. & Wang, S. Multifunctional “smart” particles engineered from live immunocytes: toward capture and release of cancer cells. Adv. Mater. 27, 310–313 (2015).
Fuster, M. M. & Esko, J. D. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat. Rev. Cancer 5, 525–542 (2005).
Park, K. Controlled drug delivery systems: past forward and future back. J. Controlled Release 190, 3–8 (2014).
Kleene, R. & Schachner, M. Glycans and neural cell interactions. Nat. Rev. Neurosci. 5, 195–208 (2004).
Palaniappan, K. K. & Bertozzi, C. R. Chemical glycoproteomics. Chem. Rev. 116, 14277–14306 (2016).
Ouyang, J., Chen, M., Bao, W.-J., Zhang, Q.-W., Wang, K. & Xia, X.-H. Morphology controlled poly(aminophenylboronic acid) nanostructures as smart substrates for enhanced capture and release of circulating tumor cells. Adv. Funct. Mater. 25, 6122–6130 (2015).
Roy, D. & Sumerlin, B. S. Glucose-sensitivity of boronic acid block copolymers at physiological pH. ACS Macro Lett. 1, 529–532 (2012).
Imperiali, B. & O’Connor, S. E. Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr. Opin. Chem. Biol. 3, 643–649 (1999).
Yabu, M., Korekane, H., Takahashi, H., Ohigashi, H., Ishikawa, O. & Miyamoto, Y. Accumulation of free Neu5Ac-containing complex-type N-glycans in human pancreatic cancers. Glycoconjugate J. 30, 247–256 (2013).
Arnaud, J., Audfray, A. & Imberty, A. Binding sugars: from natural lectins to synthetic receptors and engineered neolectins. Chem. Soc. Rev. 42, 4798–4813 (2013).
Qing, G., Lu, Q., Xiong, Y., Zhang, L., Wang, H., Li, X., Liang, X. & Sun, T. New opportunities and challenges of smart polymers in post-translational modification proteomics. Adv. Mater. 29: 1604670 (2017).
Zou, Y., Ji, X., Cai, J., Yuan, T., Stanton, D. J., Lin, Y.-H., Naraghi, M. & Fang, L. Synthesis and solution processing of a hydrogen-bonded ladder polymer. Chem 2, 9–10 (2017).
Qing, G. & Sun, T. The transformation of chiral signals into macroscopic properties of materials using chirality-responsive polymers. NPG Asia Mater. 4, e4 (2012).
Zhao, W., Wei, J.-S., Zhang, P., Chen, J., Kong, J.-L., Sun, L.-H., Xiong, H.-M. & Möhwald, H. Self-assembled ZnO nanoparticle capsules for carrying and delivering isotretinoin to cancer cells. ACS Appl. Mater. Interfaces 9, 18474–18481 (2017).
Vacca, A., Nativi, C., Cacciarini, M., Pergoli, R. & Roelens, S. A new tripodal receptor for molecular recognition of monosaccharides. A paradigm for assessing glycoside binding affinities and selectivities by 1H NMR spectroscopy. J. Am. Chem. Soc. 126, 16456–16465 (2004).
Peng, P. & Schmidt, R. R. An alternative reaction course in O-glycosilation with O-glycosyl trichloroacetimidates as glycosyl donors and Lewis acidic metal salts as catalyst: acid-base catalysis with gold chloride-glycosyl acceptor adducts. J. Am. Chem. Soc. 137, 12653–12659 (2015).
Segura, M., Bricoli, B., Casnati, A., Muñoz, E. M., Sansone, F., Ungaro, R. & Vicent, C. A prototype calix[4]arene-based receptor for carbohydrate recognition containing peptide and phosphate binding groups. J. Org. Chem. 68, 6296–6303 (2003).
Chandramouli, N., Ferrand, Y., Lautrette, G., Kauffmann, B., Mackereth, C. D., Laguerre, M., Dubreuil, D. & Huc, I. Iterative design of a helically folded aromatic oligoamide sequence for the selective encapsulation of fructose. Nat. Chem. 7, 334–341 (2015).
Wang, Q.-Q., Day, V. W. & Bowman-James, K. Chemistry and structure of a host-guest relationship: the power of NMR and X-ray diffraction in tandem. J. Am. Chem. Soc. 135, 392–399 (2013).
Zhang, K., Xu, R., Ge, W., Qi, M., Zhang, G., Xu, Q.-H., Huang, F., Cao, Y. & Wang, X. Electrostatically self-assembled chitosan derivatives working as efficient cathode interlayers for organic solar cells. Nano Energy 34, 164–171 (2017).
Liu, Q., Jin, C., Wang, Y., Fang, X., Zhang, X., Chen, Z. & Tan, W. Aptamer-conjugated nanomaterials for specific cancer cell recognition and targeted cancer therapy. NPG Asia Mater. 6, e95 (2014).
Qing, G., Li, X., Xiong, P., Chen, C., Zhan, M., Liang, X. & Sun, T. Dipeptide-based carbohydrate receptors and polymers for glycopeptide enrichment and glycan discrimination. ACS Appl. Mater. Interfaces 8, 22084–22092 (2016).
Passeri, D., Rossi, M., Tamburri, E. & Terranova, M. L. Mechanical characterization of polymeric thin films by atomic force microscopy based techniques. Anal. Bioanal. Chem. 405, 1463–1478 (2013).
Meng, X., Gui, B., Yuan, D., Zeller, M. & Wang, C. Mechanized azobenzene-functionalized zirconium metal-organic framework for on-command cargo release. Sci. Adv. 2, e1600480 (2016).
Lin, G., Ding, H., Yuan, D., Wang, B. & Wang, C. A pyrene-based, fluorescent three-dimensional covalent organic framework. J. Am. Chem. Soc. 138, 3302–3305 (2016).
Yao, X., Niu, X., Ma, K., Huang, P., Grothe, J., Kaskel, S. & Zhu, Y. Graphene quantum dots-capped magnetic mesoporous silica nanoparticles as a multifunctional platform for controlled drug delivery, magnetic hyperthermia, and photothermal therapy. Small 13, 1602225 (2017).
Tian, R., Zhang, H., Ye, M., Jiang, X., Hu, L., Li, X., Bao, X. & Zou, H. Selective extraction of peptides from human plasma by highly ordered mesoporous silica particles for peptidome analysis. Angew. Chem. Int. Ed. 46, 962–965 (2007).
Cao, Q., Zhao, X., Zhao, Q., Lv, X., Ma, C., Li, X., Zhao, Y., Peng, B., Ying, W. & Qian, X. Strategy integrating stepped fragmentation and glycan diagnostic ion-based spectrum refinement for the identification of core fucosylated glycoproteome using mass spectrometry. Anal. Chem. 86, 6804–6811 (2014).
Aronson, S. J. & Rehm, H. L. Building the foundation for genomics in precision medicine. Nature 526, 336–342 (2015).
Gunasekara, R. W. & Zhao, Y. A general method for selective recognition of monosaccharides and oligosaccharides in water. J. Am. Chem. Soc. 139, 829–835 (2017).
Noro, S. & Nakamura, T. Fluorine-functionalized metal-organic frameworks and porous coordination polymers. NPG Asia Mater. 9, e433 (2017).
Lin, G., Ding, H., Chen, R., Peng, Z., Wang, B. & Wang, C. 3D porphyrin-based covalent organic frameworks. J. Am. Chem. Soc. 139, 8705–8709 (2017).
Acknowledgements
We thank the Major State Basic Research Development Program of China (973 Program, Grant 2013CB933002), the National Natural Science Foundation of China (Grants 51533007, 51521001, 21275114, 51473131 and 21775116) and the China National Funds for Distinguished Young Scientists (Grant 51325302). GQ acknowledges the Hubei Provincial Department of Education for financial assistance through the ‘Chutian Scholar’ Program and the Hubei Provincial Natural Science Foundation of China (2014CFA039).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the NPG Asia Materials website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, Z., Lv, Z., Wang, X. et al. A biomimetic design for a sialylated, glycan-specific smart polymer. NPG Asia Mater 10, e472 (2018). https://doi.org/10.1038/am.2017.241
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2017.241
This article is cited by
-
Sensitive and specific detection of saccharide species based on fluorescence: update from 2016
Analytical and Bioanalytical Chemistry (2023)
