
News & Views |
Featured
-
-
Article |
 Stereoelectronic effects in stabilizing protein–N-glycan interactions revealed by experiment and machine learning
Stereoelectronic effects in stabilizing protein–N-glycan interactions revealed by experiment and machine learning
Analysis of the thermodynamics of protein–N-glycan interactions perturbed by mutations has revealed an enthalpy–entropy compensation that depends on the electronics of the interacting side chains. Machine-learned and statistical models showed that protein–N-glycan interactions highly correlate with stereoelectronic effects, and that a major part of protein–N-glycan interactions can be explained using the energetic rules of frontier molecular orbital interactions.
- Maziar S. Ardejani
- , Louis Noodleman
- & Jeffery W. Kelly
-
Article |
 Optimization of the facet structure of transition-metal catalysts applied to the oxygen reduction reaction
Optimization of the facet structure of transition-metal catalysts applied to the oxygen reduction reaction
While much effort has been devoted to understanding how nanoparticle morphology can be leveraged to improve catalytic activity, engineering their microstructure from first principles to this end has remained difficult. Now a methodology for designing the optimal structure of a solid catalyst with the aim of achieving the highest possible activity for surface-sensitive reactions has been developed.
- M. Núñez
- , J. L. Lansford
- & D. G. Vlachos
-
News & Views |
 Encoding evolution of porous solids
Encoding evolution of porous solids
The design and prediction of network topology is challenging, even when the components' principle interactions are strong. Now, frameworks with relatively weak 'chiral recognition' between organic building blocks have been synthesized and rationalized in silico — an important development in the reticular synthesis of molecular crystals.
- Caroline Mellot-Draznieks
- & Anthony K. Cheetham
-
Article |
 Reticular synthesis of porous molecular 1D nanotubes and 3D networks
Reticular synthesis of porous molecular 1D nanotubes and 3D networks
Porous molecular crystals have desirable properties, but are hard to form with the level of structural control seen for extended framework materials. Now, a ‘mix-and-match’ chiral recognition strategy has been used to form reticular porous supramolecular nanotubes and 3D networks, providing a blueprint for pore design in molecular crystals.
- A. G. Slater
- , M. A. Little
- & A. I. Cooper
-
News & Views |
 Fixing Jacob's ladder
Fixing Jacob's ladder
Density functional theory calculations can be carried out with different levels of accuracy, forming a hierarchy that is often represented by the rungs of a ladder. Now a new method has been developed that significantly improves the accuracy of the 'third rung' when calculating the properties of diversely bonded systems.
- Roberto Car
-
Article |
 Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional
Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional
Whether a molecule or material can exist, and with what structures and energies, is of critical importance. For demanding calculations the efficiency of density functional theory makes it the only practical electronic structure theory available to help answer these questions. Now, an efficient density functional is shown to have unprecedented accuracy for a diverse set of bonded systems.
- Jianwei Sun
- , Richard C. Remsing
- & John P. Perdew
-
Article |
 Synthesis and stability of xenon oxides Xe2O5 and Xe3O2 under pressure
Synthesis and stability of xenon oxides Xe2O5 and Xe3O2 under pressure
The reactivity of the noble gases—a notoriously inert group—at high pressures is intriguing. Now, two xenon oxides with unusual stoichiometries, Xe2O5 and Xe3O2, have been synthesized above 78 GPa and predicted to be stable above 50 GPa, indicating that xenon is more reactive than previously thought.
- Agnès Dewaele
- , Nicholas Worth
- & Tetsuo Irifune
-
News & Views |
 The quest for new functionality
The quest for new functionality
Building on our understanding of the chemical bond, advances in synthetic chemistry, and large-scale computation, materials design has now become a reality. From a pool of 400 unknown compositions, 15 new compounds have been realized that adopt the predicted structures and properties.
- Aron Walsh
-
Article |
 Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds
Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds
A method to predict the stability, structure and properties of as-yet-unreported materials has been devised. For 18-valence electron ABX materials, 15 such ‘missing’ compounds identified to be thermodynamically stable were successfully synthesized, and showed crystal structures and properties in good agreement with the predicted ones.
- Romain Gautier
- , Xiuwen Zhang
- & Alex Zunger
-
Article |
 Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels
Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels
Peptides that self-assemble into nanostructures are of interest for many applications, including ones relevant to cosmetics, food, biomedicine and nanotechnology. Now, computational tools have been developed that enable peptide sequence space to be rapidly searched for supramolecular properties and this approach has been used to identify unprotected tripeptide hydrogelators.
- Pim W. J. M. Frederix
- , Gary G. Scott
- & Tell Tuttle
-
News & Views |
 A virtual squeeze on chemistry
A virtual squeeze on chemistry
Molecular simulations have the potential to give valuable insights into experimental results, but can be limited by the time- and length-scales they can simulate. Now, reactive chemistry can be driven through a novel simulation approach, which could have ramifications for many research areas, including astrobiology and the origins of life.
- Nir Goldman
-
Article |
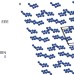 Calculations predict a stable molecular crystal of N8
Calculations predict a stable molecular crystal of N8
Polynitrogen compounds are of interest on a fundamental level and as potential high-energy-density materials. A crystalline solid that consists of two isomeric forms of N8 molecules held together by weak van der Waals interactions has now been predicted to exist, and to be stable even at low pressures.
- Barak Hirshberg
- , R. Benny Gerber
- & Anna I. Krylov
-
News & Views |
 Beyond state I
Beyond state I
Although caesium is well known in its oxidation state +I, many chemists have speculated about a possible higher state. Such a species has not yet been prepared, but based on quantum-chemical calculations CsFn compounds have now been predicted to be stable.
- Sebastian Riedel
- & Peter Schwerdtfeger
-
Article |
 Caesium in high oxidation states and as a p-block element
Caesium in high oxidation states and as a p-block element
Caesium has so far not been found in oxidation states higher than +1, but quantum chemical calculations have now shown that, under high pressures, 5p inner shell electrons of caesium can participate in — and become the main components of — bonds. Caesium is predicted to form stable CsFn molecules that resemble isoelectronic XeFn.
- Mao-sheng Miao
 Fast track to structural biology
Fast track to structural biology