
Article
|
Open Access
Featured
-
-
Article
| Open Access Genome and epigenome wide studies of neurological protein biomarkers in the Lothian Birth Cohort 1936
Genome and epigenome wide studies of neurological protein biomarkers in the Lothian Birth Cohort 1936
Plasma levels of neurological proteins have the potential to serve as biomarkers for neurological conditions. Here, Hillary et al. perform genome- and epigenome-wide association studies for 92 neurological proteins and identify 41 genomic loci for 33 proteins and 26 CpG sites for 9 proteins.
- Robert F. Hillary
- , Daniel L. McCartney
- & Riccardo E. Marioni
-
Article
| Open Access Reconstitution of recombinant human CCR4-NOT reveals molecular insights into regulated deadenylation
Reconstitution of recombinant human CCR4-NOT reveals molecular insights into regulated deadenylation
The CCR4-NOT complex shortens poly(A) tails of messenger RNAs. By biochemical reconstitution of the entire human CCR4-NOT complex, the authors show the stimulatory roles of non-enzymatic subunits and the importance of the interaction between CAF40 and RNA binding proteins in targeted deadenylation.
- Tobias Raisch
- , Chung-Te Chang
- & Eugene Valkov
-
Article
| Open Access Empirical mean-noise fitness landscapes reveal the fitness impact of gene expression noise
Empirical mean-noise fitness landscapes reveal the fitness impact of gene expression noise
Quantifying the effects of noise in gene expression is difficult since noise and mean expression are coupled. Here the authors determine fitness landscapes in mean-noise expression space to uncouple these two parameters and show that changes in noise and mean expression are similarly detrimental to fitness.
- Jörn M. Schmiedel
- , Lucas B. Carey
- & Ben Lehner
-
Article
| Open Access Hox11 expressing regional skeletal stem cells are progenitors for osteoblasts, chondrocytes and adipocytes throughout life
Hox11 expressing regional skeletal stem cells are progenitors for osteoblasts, chondrocytes and adipocytes throughout life
Prior evidence suggested mesenchymal stromal cells (MSCs) required for skeletal formation, maintenance, and repair arise postnatally. Here, the authors show that Hoxa11 lineage-marked cells give rise to all skeletal lineages from embryogenesis through adulthood and are upstream progenitors of LepR- and Osx-lineage MSCs
- Kyriel M. Pineault
- , Jane Y. Song
- & Deneen M. Wellik
-
Article
| Open Access Systematic allelic analysis defines the interplay of key pathways in X chromosome inactivation
Systematic allelic analysis defines the interplay of key pathways in X chromosome inactivation
Xist RNA is the master regulator of X chromosome inactivation. Here the authors describe a systematic analysis of Xist-mediated allelic silencing in mouse ESC models and define the contribution of different pathways that regulate gene silencing.
- Tatyana B. Nesterova
- , Guifeng Wei
- & Neil Brockdorff
-
Article
| Open Access Zebrafish preserve global germline DNA methylation while sex-linked rDNA is amplified and demethylated during feminisation
Zebrafish preserve global germline DNA methylation while sex-linked rDNA is amplified and demethylated during feminisation
Germline cells transfer genetic information to offspring, and in zebrafish, drive sex determination. Here the authors report that, unlike mammals, the germline of zebrafish does not undergo genome-wide DNA methylation erasure, while amplifying and demethylating sex-linked rDNA during feminisation.
- Oscar Ortega-Recalde
- , Robert C. Day
- & Timothy A. Hore
-
Article
| Open Access Detection of cell-type-specific risk-CpG sites in epigenome-wide association studies
Detection of cell-type-specific risk-CpG sites in epigenome-wide association studies
Cellular heterogeneity is one of the major confounding factors in EWAS studies. Here the authors present a statistical method, HIgh REsolution (HIRE), which enables the detection of risk-CpG sites for individual cell types.
- Xiangyu Luo
- , Can Yang
- & Yingying Wei
-
Article
| Open Access Mutation bias and GC content shape antimutator invasions
Mutation bias and GC content shape antimutator invasions
Mutators are expected to re-evolve low mutation rates to reduce deleterious load, but empirical evidence is mixed. Here, the authors show that load can vary across mutators and genetic backgrounds, which their simulations suggest can substantially alter antimutator dynamics.
- Alejandro Couce
- & Olivier Tenaillon
-
Article
| Open Access Human DEF6 deficiency underlies an immunodeficiency syndrome with systemic autoimmunity and aberrant CTLA-4 homeostasis
Human DEF6 deficiency underlies an immunodeficiency syndrome with systemic autoimmunity and aberrant CTLA-4 homeostasis
CTLA-4 is critical for balancing protective immunity with self-tolerance. Here the authors identify homozygous DEF6 mutations in patients with severe autoimmunity, one of which received and responds to CTLA-4-Ig, and show that DEF6 is crucial for CTLA-4 cell surface trafficking and immune regulatory function.
- Nina K. Serwas
- , Birgit Hoeger
- & Kaan Boztug
-
Article
| Open Access Association of imputed prostate cancer transcriptome with disease risk reveals novel mechanisms
Association of imputed prostate cancer transcriptome with disease risk reveals novel mechanisms
In prostate cancer, investigating aberrant gene expression may shed light on disease etiology. Here, the authors imputed expression transcriptome-wide for 233,955 European ancestry men, discovering and replicating the associations between prostatic expression for select genes and prostate cancer risk, including the highly prevalent gene fusion partner TMPRSS2. The authors furthermore integrate diverse functional genomic datasets to interpret the epigenetic mechanisms by which the implicated risk variants and genes modulate disease risk.
- Nima C. Emami
- , Linda Kachuri
- & John S. Witte
-
Article
| Open Access AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders
AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders
Genetic variants in ionotropic glutamate receptors have been implicated in neurodevelopmental disorders. Here, the authors report heterozygous de novo mutations in the GRIA2 gene in 28 individuals with intellectual disability and neurodevelopmental abnormalities associated with reduced Ca2+ transport and AMPAR currents.”
- Vincenzo Salpietro
- , Christine L. Dixon
- & Henry Houlden
-
Article
| Open Access Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation
Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation
The role of brain somatic mutations in neurodegenerative diseases such as Alzheimer’s disease (AD) is not well understood. Here the authors carry out high-depth exome sequencing ~500× on brain tissue from patients with AD and controls, and identify mutations in a number of genes that are known to contribute to phosphorylation and aggregation of tau, including PIN1.
- Jun Sung Park
- , Junehawk Lee
- & Jeong Ho Lee
-
Article
| Open Access The nasal methylome as a biomarker of asthma and airway inflammation in children
The nasal methylome as a biomarker of asthma and airway inflammation in children
Epigenetic differences in nasal epithelium have been proposed as a biomarker for lower airway disease and asthma. Here, in epigenome-wide association studies for asthma and other airway traits using nasal swabs, the authors identify differentially methylated CpGs that highlight genes involved in TH2 response.
- Andres Cardenas
- , Joanne E. Sordillo
- & Diane R. Gold
-
Article
| Open Access Pioneer and repressive functions of p63 during zebrafish embryonic ectoderm specification
Pioneer and repressive functions of p63 during zebrafish embryonic ectoderm specification
The transcription factor p63 is a master regulator of ectoderm development. Here the authors show that zebrafish p63 binds enhancers associated with neural genes to limit Sox3 binding and gene expression while also acting as a pioneer factor by promoting chromatin opening at epidermal gene enhancers.
- José M. Santos-Pereira
- , Lourdes Gallardo-Fuentes
- & Juan J. Tena
-
Article
| Open Access Retention of paternal DNA methylome in the developing zebrafish germline
Retention of paternal DNA methylome in the developing zebrafish germline
Germ cells are the means of transferring genetic information to the next generation. Here the authors characterise the DNA methylomes of zebrafish primordial germ cells and find that, unlike mammals, the zebrafish germ cells do not undergo genome-wide DNA demethylation but rather retain paternal DNA methylation patterns
- Ksenia Skvortsova
- , Katsiaryna Tarbashevich
- & Ozren Bogdanovic
-
Article
| Open Access Paternal-age-related de novo mutations and risk for five disorders
Paternal-age-related de novo mutations and risk for five disorders
Advanced paternal age associates with increased risk for psychiatric and developmental disorders in offspring. Here, Taylor et al. utilize parent-child trio exome sequencing data sets to estimate the contribution of paternal age-related de novo mutations to multiple disorders, including heart disease and schizophrenia.
- Jacob L. Taylor
- , Jean-Christophe P. G. Debost
- & Elise B. Robinson
-
Article
| Open Access A genome-wide scan statistic framework for whole-genome sequence data analysis
A genome-wide scan statistic framework for whole-genome sequence data analysis
Whole-genome sequencing data reveals a large number of variants for testing their associations with phenotypic traits and diseases. Here, the authors develop WGScan, a statistical method for detecting the existence and estimating the locations of the association signal at genome-wide scale.
- Zihuai He
- , Bin Xu
- & Iuliana Ionita-Laza
-
Article
| Open Access Mapping the drivers of within-host pathogen evolution using massive data sets
Mapping the drivers of within-host pathogen evolution using massive data sets
Various host factors may impact within-host pathogen evolution. Here, the authors develop a Bayesian approach for identifying host-pathogen interactions using large data sets of pathogen diversity, and apply it to investigate HLA-induced selection in the HIV-1 genome.
- Duncan S. Palmer
- , Isaac Turner
- & Gil McVean
-
Article
| Open Access Monoallelic expression and epigenetic inheritance sustained by a Trypanosoma brucei variant surface glycoprotein exclusion complex
Monoallelic expression and epigenetic inheritance sustained by a Trypanosoma brucei variant surface glycoprotein exclusion complex
Monoallelic expression of variant surface glycoprotein genes (VSGs) is essential for immune evasion by Trypanosoma brucei. Here, Faria et al. show that the VEX protein complex controls VSG allelic exclusion, and that CAF‐1 sustains inheritance of the VEX‐complex in association with the active VSG.
- Joana Faria
- , Lucy Glover
- & David Horn
-
Article
| Open Access The effect of X-linked dosage compensation on complex trait variation
The effect of X-linked dosage compensation on complex trait variation
Dosage compensation (DC) on the X chromosome has predictable effects on genetic and phenotypic trait variance. Here, the authors use information for 20 quantitative traits in the UK Biobank and across-tissue gene expression to compare X-linked heritability and the effects of trait-associated SNPs between the sexes.
- Julia Sidorenko
- , Irfahan Kassam
- & Peter M. Visscher
-
Article
| Open Access Chemical genomics reveals histone deacetylases are required for core regulatory transcription
Chemical genomics reveals histone deacetylases are required for core regulatory transcription
Core regulatory transcription factors are usually regulated by cell-type specific super enhancers (SEs). Here, the authors screen for chemical probes able to distinguish between SE-driven and promoter-driven transcription and find that histone deacetylases are selectively required for core regulatory transcription.
- Berkley E. Gryder
- , Lei Wu
- & Javed Khan
-
Article
| Open Access Clinically-relevant postzygotic mosaicism in parents and children with developmental disorders in trio exome sequencing data
Clinically-relevant postzygotic mosaicism in parents and children with developmental disorders in trio exome sequencing data
Systematic analysis of postzygotic mosaicism (PZM) is difficult due to challenges in detecting such events. Here, Wright et al. analyse trio exome sequencing data from blood and saliva of 4,293 probands with developmental disorders from the DDD Study and estimate that >3% of causative de novo mutations result from PZM.
- C. F. Wright
- , E. Prigmore
- & M. E. Hurles
-
Article
| Open Access Extensive intraspecific gene order and gene structural variations in upland cotton cultivars
Extensive intraspecific gene order and gene structural variations in upland cotton cultivars
While multiple cotton genomes are available, genome wide variation comparison between allotetraploid upland cotton cultivars remain unexplored. Here, the authors assemble two upland cotton cultivars and reveal large scale structural variations on chromosome A08.
- Zhaoen Yang
- , Xiaoyang Ge
- & Fuguang Li
-
Article
| Open Access Genomic signatures and correlates of widespread population declines in salmon
Genomic signatures and correlates of widespread population declines in salmon
The Atlantic salmon has suffered widespread population declines over the last century. Here, Lehnert et al. reconstruct changes in effective population size of 172 populations based on genomic linkage information revealing mostly temperature-associated population declines with over 60% of populations in decline since 1975.
- S. J. Lehnert
- , T. Kess
- & I. R. Bradbury
-
Article
| Open Access Target preference of Type III-A CRISPR-Cas complexes at the transcription bubble
Target preference of Type III-A CRISPR-Cas complexes at the transcription bubble
Type III CRISPR-Cas systems are able to target transcriptionally active DNA sequences in phages and plasmids. Here, the authors reveal the mechanism of the target nucleic acid preference of Type III-A CRISPR-Cas complexes at the transcription bubble by a combination of structural and biochemical approaches.
- Tina Y. Liu
- , Jun-Jie Liu
- & Jennifer A. Doudna
-
Article
| Open Access Dissecting a heterotic gene through GradedPool-Seq mapping informs a rice-improvement strategy
Dissecting a heterotic gene through GradedPool-Seq mapping informs a rice-improvement strategy
Developing hybrid rice cultivars requires time consuming random crossing. Here, the authors develop a new next generation sequencing-based quantitative trait locus mapping method to dissect heterotic gene OsMADS1 and demonstrate the feasibility of pyramiding two genes to achieve large heterotic effect.
- Changsheng Wang
- , Shican Tang
- & Bin Han
-
Article
| Open Access Accurate estimation of cell-type composition from gene expression data
Accurate estimation of cell-type composition from gene expression data
Bulk RNA-seq data harbors valuable information about gene expression levels from different cell types in tissue samples. Here, the authors develop DWLS, a computational method for estimating cell-type composition of bulk data by leveraging single-cell RNA-seq-derived cell-type signatures.
- Daphne Tsoucas
- , Rui Dong
- & Guo-Cheng Yuan
-
Article
| Open Access A practical guide for mutational signature analysis in hematological malignancies
A practical guide for mutational signature analysis in hematological malignancies
Mutational signature analysis provides important information about the mutational processes underpinning different stages of tumorigenesis. Here, the authors compare publicly available signature extraction tools and suggest a framework for the generation of accurate and reproducible signature data.
- Francesco Maura
- , Andrea Degasperi
- & Niccolò Bolli
-
Article
| Open Access The transcription factor OsSUF4 interacts with SDG725 in promoting H3K36me3 establishment
The transcription factor OsSUF4 interacts with SDG725 in promoting H3K36me3 establishment
The distribution of H3K36me3 varies between species. Here Liu et al. show that the OsSUF4 transcription factor binds its target motif via a zinc finger domain to promote H3K36 methyltransferase targeting close to the transcription start site of genes including the flowering regulators RFT1 and Hd3a.
- Bing Liu
- , Yuhao Liu
- & Aiwu Dong
-
Article
| Open Access Mutations in SMARCB1 and in other Coffin–Siris syndrome genes lead to various brain midline defects
Mutations in SMARCB1 and in other Coffin–Siris syndrome genes lead to various brain midline defects
Why and how mutations in genes encoding BAF complex components lead to distinct disease entitites remains unresolved. In this study, authors establish the first Smarcb1 mutant mouse model with multiple brain abnormalities recapitulating human Coffin–Siris syndrome and show that one prominent midline abnormality, corpus callosum agenesis, is due to midline glia aberrations.
- Alina Filatova
- , Linda K. Rey
- & Ulrike A. Nuber
-
Article
| Open Access Mendelian randomisation analysis of the effect of educational attainment and cognitive ability on smoking behaviour
Mendelian randomisation analysis of the effect of educational attainment and cognitive ability on smoking behaviour
Higher educational attainment is positively associated with a number of health outcomes. Here, Sanderson et al. use multivariable Mendelian randomisation analysis to test whether the association of educational attainment with smoking behaviour is direct or indirectly mediated via general cognitive ability.
- Eleanor Sanderson
- , George Davey Smith
- & Marcus R. Munafò
-
Article
| Open Access PRC1 collaborates with SMCHD1 to fold the X-chromosome and spread Xist RNA between chromosome compartments
PRC1 collaborates with SMCHD1 to fold the X-chromosome and spread Xist RNA between chromosome compartments
The inactive X (Xi)-specific S1/S2 chromosome compartments are merged by SMCHD1, but how the S1/S2 structure is constructed is unclear. The authors find that PRC1 drives the formation of S1/S2s and that the stepwise folding process of the Xi facilitates Xist RNA spreading between Xi compartments.
- Chen-Yu Wang
- , David Colognori
- & Jeannie T. Lee
-
Article
| Open Access E2F4 regulates transcriptional activation in mouse embryonic stem cells independently of the RB family
E2F4 regulates transcriptional activation in mouse embryonic stem cells independently of the RB family
E2F transcription factors are regulators of cell division and cell fate decisions. Here the authors show that E2F4 is important for proliferation and survival of mouse ESCs, independent of the RB family, and that E2F4 interacts with chromatin regulators associated with gene activation.
- Jenny Hsu
- , Julia Arand
- & Julien Sage
-
Article
| Open Access Nuclei multiplexing with barcoded antibodies for single-nucleus genomics
Nuclei multiplexing with barcoded antibodies for single-nucleus genomics
Single-nucleus RNA-seq enables interrogation of complex tissues but is limited due to batch effects and processing costs. Here the authors use barcoded antibodies against the nuclear pore complex to label nuclei from distinct samples, and develop a computational tool to assign the sample of origin.
- Jellert T. Gaublomme
- , Bo Li
- & Aviv Regev
-
Article
| Open Access Enhancer accessibility and CTCF occupancy underlie asymmetric TAD architecture and cell type specific genome topology
Enhancer accessibility and CTCF occupancy underlie asymmetric TAD architecture and cell type specific genome topology
Eukaryotic genomes fold into topologically associated domains (TAD). Here the authors characterise a TAD regulatory architecture underlying lineage-specific gene regulation, finding that stripe TADs are associated with poised and active chromatin landscapes and linked to the cells functional state.
- Christopher Barrington
- , Dimitra Georgopoulou
- & Suzana Hadjur
-
Article
| Open Access Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napus
Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napus
Despite the economic importance of the allotetraploid crop Brassica napus, our knowledge of its phylogenomic relationships, genetic structure, and diversification is limited. Here, the authors show its complex origin and diversification by analyzing transcriptome and organellar sequencing data.
- Hong An
- , Xinshuai Qi
- & J. Chris Pires
-
Article
| Open Access Improving the diagnostic yield of exome- sequencing by predicting gene–phenotype associations using large-scale gene expression analysis
Improving the diagnostic yield of exome- sequencing by predicting gene–phenotype associations using large-scale gene expression analysis
A genetic diagnosis remains unattainable for many individuals with a rare disease because of incomplete knowledge about the genetic basis of many diseases. Here, the authors present the web-based tool GADO (GeneNetwork Assisted Diagnostic Optimization) that uses public RNA-seq data for prioritization of candidate genes.
- Patrick Deelen
- , Sipko van Dam
- & Lude Franke
-
Article
| Open Access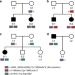 A frequent variant in the Japanese population determines quasi-Mendelian inheritance of rare retinal ciliopathy
A frequent variant in the Japanese population determines quasi-Mendelian inheritance of rare retinal ciliopathy
Genetic variants in RP1 can cause hereditary retinal degeneration (HRD). Here, in a genomic screen of 331 Japanese HRD patients, the authors identify a near-polymorphic RP1 variant that causes Mendelian HRD in trans with an Alu insertion and otherwise is associated with HRD according to a complex model of inheritance.
- Konstantinos Nikopoulos
- , Katarina Cisarova
- & Carlo Rivolta
-
Article
| Open Access Chromatin organization in the female mouse brain fluctuates across the oestrous cycle
Chromatin organization in the female mouse brain fluctuates across the oestrous cycle
The molecular mechanisms underlying the dynamic nature of the female brain structure and function remain poorly understood. Here the authors characterise chromatin organization in the mouse female ventral hippocampus, finding it fluctuates with the oestrous cycle, and identify changes in chromatin organization associated with the transcription of genes important for neuronal function and behaviour.
- Ivana Jaric
- , Devin Rocks
- & Marija Kundakovic
-
Article
| Open Access Efficient base editing for multiple genes and loci in pigs using base editors
Efficient base editing for multiple genes and loci in pigs using base editors
Base editors can efficiently produce single nucleotide alterations without requiring a double-strand break. Here the authors show base editing at multiple sites simultaneously in pigs.
- Jingke Xie
- , Weikai Ge
- & Liangxue Lai
-
Article
| Open Access Common and distinct transcriptional signatures of mammalian embryonic lethality
Common and distinct transcriptional signatures of mammalian embryonic lethality
The transcriptional signature of embryonic lethality has not been defined. Here, the authors, as part of the Deciphering the Mechanisms of Developmental Disorders programme, define genes causing murine embryonic lethality around E9.5 and identify developmental delay transcriptional signatures.
- John E. Collins
- , Richard J. White
- & Elisabeth M. Busch-Nentwich
-
Article
| Open Access DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation
DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation
Histone 3 lysine 79 is mono (me1), di (me2), or tri (me3) methylated by the methyltransferase DOT1L. Here the authors reveal a group of enhancers defined by H3K79me2/3 which regulates enhancer-promoter interactions and other key enhancer features in MLL-AF4 leukemia cells.
- Laura Godfrey
- , Nicholas T. Crump
- & Thomas A. Milne
-
Article
| Open Access Genome-wide analysis of dental caries and periodontitis combining clinical and self-reported data
Genome-wide analysis of dental caries and periodontitis combining clinical and self-reported data
Dental caries and periodontitis are among the most common medical conditions. Here, the authors report a GWAS for measures of oral health that reveals 47 risk loci for caries, find genetic correlation with 31 other complex traits and use Mendelian randomization analyses to explore potential causal relationships.
- Dmitry Shungin
- , Simon Haworth
- & Ingegerd Johansson
-
Article
| Open Access Chromosome dynamics near the sol-gel phase transition dictate the timing of remote genomic interactions
Chromosome dynamics near the sol-gel phase transition dictate the timing of remote genomic interactions
Antibodies are generated through remote genomic interactions involving immunoglobulin variable (VH), diversity (DH) and joining (JH) gene segments. Here the authors develop a strategy to track VH-DHJH motion in B-lymphocytes and provide evidence that chromosome organisation near the sol-gel phase transition dictates the timing of genomic interactions to orchestrate gene expression and somatic recombination.
- Nimish Khanna
- , Yaojun Zhang
- & Cornelis Murre
-
Article
| Open Access Pathologic gene network rewiring implicates PPP1R3A as a central regulator in pressure overload heart failure
Pathologic gene network rewiring implicates PPP1R3A as a central regulator in pressure overload heart failure
The genetic and pathogenetic basis of heart failure is incompletely understood. Here, the authors present a high-fidelity tissue collection from rapidly preserved failing and non-failing control hearts which are used for eQTL mapping and network analysis, resulting in the prioritization of PPP1R3A as a heart failure gene.
- Pablo Cordero
- , Victoria N. Parikh
- & Euan A. Ashley
-
Article
| Open Access HIV-1 DNA sequence diversity and evolution during acute subtype C infection
HIV-1 DNA sequence diversity and evolution during acute subtype C infection
The dynamics of HIV-1 DNA sequences early after HIV-1 transmission remains poorly characterized. Here, the authors perform a longitudinal evaluation of HIV-1 DNA sequences in subtype C-infected individuals during acute infection, providing a landscape of the nature and evolution of the very early viral genome.
- Guinevere Q. Lee
- , Kavidha Reddy
- & Mathias Lichterfeld
-
Article
| Open Access Integrative inference of subclonal tumour evolution from single-cell and bulk sequencing data
Integrative inference of subclonal tumour evolution from single-cell and bulk sequencing data
Intra-tumour heterogeneity provides important information about subclonal tumour evolution. Here, the authors develop B-SCITE, a computational method for inferring tumour phylogenies from combined single-cell and bulk sequencing data.
- Salem Malikic
- , Katharina Jahn
- & Niko Beerenwinkel
-
Article
| Open Access Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia
Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia
There is increasing evidence that epigenetic mechanisms contribute to therapeutic resistance in cancer. Here the authors study AML patient samples and a mouse model of non-genetic resistance and find that transcriptional plasticity drives stable epigenetic resistance, and identify regulators of enhancer function as important modulators of resistance.
- Charles C. Bell
- , Katie A. Fennell
- & Mark A. Dawson
-
Article
| Open Access The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposable elements in Paramecium
The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposable elements in Paramecium
H3K9me3 and H3K27me3 chromatin silencing marks are usually deposited by different SET-domain proteins. Here the authors show that the Enhancer-of-zeste-like protein Ezl1, from the unicellular eukaryote Paramecium tetraurelia, catalyzes methylation of histone H3 in vitro and in vivo with an apparent specificity toward K9 and K27, and controls the repression of transposable elements.
- Andrea Frapporti
- , Caridad Miró Pina
- & Sandra Duharcourt
 Alternative splicing regulates stochastic NLRP3 activity
Alternative splicing regulates stochastic NLRP3 activityBrowse broader subjects
Browse narrower subjects
- Agricultural genetics
- Animal breeding
- Behavioural genetics
- Cancer genetics
- Cancer genomics
- Clinical genetics
- Consanguinity
- CRISPR-Cas systems
- Cytogenetics
- Development
- Epigenetics
- Epigenomics
- Eukaryote
- Evolutionary biology
- Functional genomics
- Gene expression
- Gene regulation
- Genetic association study
- Genetic hybridization
- Genetic interaction
- Genetic linkage study
- Genetic markers
- Genome
- Genomic instability
- Genomics
- Genotype
- Haplotypes
- Heritable quantitative trait
- Immunogenetics
- Inbreeding
- Medical genetics
- Microbial genetics
- Mutation
- Neurodevelopmental disorders
- Plant breeding
- Plant genetics
- Polyploidy
- Population genetics
- Prokaryote
- Quantitative trait
- RNA splicing
- RNAi
- Sequencing
