
Featured
-
-
Article
| Open Access InPACT: a computational method for accurate characterization of intronic polyadenylation from RNA sequencing data
InPACT: a computational method for accurate characterization of intronic polyadenylation from RNA sequencing data
Intronic polyadenylation (IPA) can produce transcripts with truncated coding regions and has been implicated in diverse biological processes and diseases. Here, the authors present a computational method for the accurate delineation of IPA events using RNA-sequencing data.
- Xiaochuan Liu
- , Hao Chen
- & Yang Yang
-
Article
| Open Access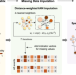 Interrogations of single-cell RNA splicing landscapes with SCASL define new cell identities with physiological relevance
Interrogations of single-cell RNA splicing landscapes with SCASL define new cell identities with physiological relevance
RNA splicing serves as a critical layer of gene expression regulation. Here, authors introduce SCASL for investigating the heterogeneity of RNA splicing landscapes at single-cell resolution, offering a novel scheme for classifying cell identities with physiological relevance.
- Xianke Xiang
- , Yao He
- & Xuerui Yang
-
Article
| Open Access A distinct class of pan-cancer susceptibility genes revealed by an alternative polyadenylation transcriptome-wide association study
A distinct class of pan-cancer susceptibility genes revealed by an alternative polyadenylation transcriptome-wide association study
Alternative polyadenylation (APA) can play a key role in cancer initiation and progression. Here, the authors conducted a comprehensive pan-cancer APA TWAS analysis and discovered a distinct class of APA-mediated cancer susceptibility genes across 22 cancer types.
- Hui Chen
- , Zeyang Wang
- & Lei Li
-
Article
| Open Access scCASE: accurate and interpretable enhancement for single-cell chromatin accessibility sequencing data
scCASE: accurate and interpretable enhancement for single-cell chromatin accessibility sequencing data
Single-cell chromatin accessibility sequencing (scCAS) data suffers from high sparsity and dimensionality. Here, authors propose an accurate and interpretable computational framework for enhancing scCAS data that considers cell-to-cell similarity.
- Songming Tang
- , Xuejian Cui
- & Shengquan Chen
-
Article
| Open Access Accurate global and local 3D alignment of cryo-EM density maps using local spatial structural features
Accurate global and local 3D alignment of cryo-EM density maps using local spatial structural features
Density map alignment is a fundamental step in Cryo-EM data postprocessing. Here, authors propose an accurate global and local density map alignment method using local density features.
- Bintao He
- , Fa Zhang
- & Renmin Han
-
Article
| Open Access Multi-omics analysis in human retina uncovers ultraconserved cis-regulatory elements at rare eye disease loci
Multi-omics analysis in human retina uncovers ultraconserved cis-regulatory elements at rare eye disease loci
Ultraconserved non-coding elements (UCNEs) can regulate developmental gene expression. Retinal multi-omics data integration revealed UCNEs to be candidate cis-regulatory elements during retinal development, which may be implicated in rare eye diseases.
- Victor Lopez Soriano
- , Alfredo Dueñas Rey
- & Elfride De Baere
-
Article
| Open Access Efficient encoding of large antigenic spaces by epitope prioritization with Dolphyn
Efficient encoding of large antigenic spaces by epitope prioritization with Dolphyn
Profiling antibody responses to vast antigenic spaces has been challenging using programmable phage display (PhIP-Seq). Here, authors develop a methodology for compressing large proteomic spaces and have discovered human antibodies targeting gut bacteria-infecting phages.
- Anna-Maria Liebhoff
- , Thiagarajan Venkataraman
- & H. Benjamin Larman
-
Article
| Open Access Recurrent evolutionary switches of mitochondrial cytochrome c maturation systems in Archaeplastida
Recurrent evolutionary switches of mitochondrial cytochrome c maturation systems in Archaeplastida
Cytochrome c maturation (CCM) is the process of covalent attachment of a heme group to the conserved cysteines to form the holocytochrome. Here, the authors report that the non-adaptive convergent evolution at the pathway level leads to mosaic distribution of CCM systems I and III among Archaeplastida species.
- Huang Li
- , Soujanya Akella
- & Jeffrey P. Mower
-
Article
| Open Access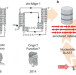 Identification of HDV-like theta ribozymes involved in tRNA-based recoding of gut bacteriophages
Identification of HDV-like theta ribozymes involved in tRNA-based recoding of gut bacteriophages
The diverse functional roles of ribozymes (RNAs with enzymatic activity) continue to be uncovered. Here, the authors identify and characterize a subgroup of minimal hepatitis delta virus (HDV)-like ribozymes – termed Theta ribozymes -, which they show process viral tRNA transcripts, and appearing crucial for lytic gene expression in recoded phages.
- Kasimir Kienbeck
- , Lukas Malfertheiner
- & Roland K. O. Sigel
-
Article
| Open Access A genome and gene catalog of the aquatic microbiomes of the Tibetan Plateau
A genome and gene catalog of the aquatic microbiomes of the Tibetan Plateau
The Tibetan Plateau is the largest plateau in the world and hosts a variety of aquatic ecosystems. Here, the authors present a gene and genome catalogue of Tibetan Plateau aquatic microbiomes, greatly expanding known taxonomic and functional diversity for the region and giving insights into its microbial biogeography.
- Mingyue Cheng
- , Shuai Luo
- & Kang Ning
-
Article
| Open Access SiFT: uncovering hidden biological processes by probabilistic filtering of single-cell data
SiFT: uncovering hidden biological processes by probabilistic filtering of single-cell data
Cells simultaneously encode multiple signals, some harder to recover. Here, authors introduce SiFT (Signal FilTering), a kernel-based projection method, revealing underlying biological processes in single-cell data.
- Zoe Piran
- & Mor Nitzan
-
Article
| Open Access High-throughput deconvolution of 3D organoid dynamics at cellular resolution for cancer pharmacology with Cellos
High-throughput deconvolution of 3D organoid dynamics at cellular resolution for cancer pharmacology with Cellos
Computational methods to analyse 3D organoids in high-throughput and with high cellular resolution remain scarce. Here, the authors propose Cellos, a high-throughput pipeline for 3D organoid segmentation using classical algorithms and a trained convolutional neural network.
- Patience Mukashyaka
- , Pooja Kumar
- & Jeffrey H. Chuang
-
Article
| Open Access Gut microbial structural variation associates with immune checkpoint inhibitor response
Gut microbial structural variation associates with immune checkpoint inhibitor response
Here, using datasets from the gut microbiome of 996 patients from seven clinical trials, the authors characterize gut microbial genomic structural variants, located in species such as Akkermansia muciniphila, Dorea formicigenerans, and Bacteroides caccae, that associate with hosts’ response and survival after immune checkpoint inhibitors treatment.
- Rong Liu
- , You Zou
- & Dao-Ming Wang
-
Article
| Open Access Dimension-agnostic and granularity-based spatially variable gene identification using BSP
Dimension-agnostic and granularity-based spatially variable gene identification using BSP
Identifying spatially variable genes (SVGs) is essential for linking molecular cell functions with tissue phenotypes. Here, authors introduce a non-parametric model that detects SVGs from two or three-dimensional spatial transcriptomics data by comparing gene expression patterns at granularities.
- Juexin Wang
- , Jinpu Li
- & Dong Xu
-
Article
| Open Access Spatial-linked alignment tool (SLAT) for aligning heterogenous slices
Spatial-linked alignment tool (SLAT) for aligning heterogenous slices
Spatial omics technologies reveal the organisation of cells in various biological systems. Here, authors propose SLAT, a graph-based algorithm for aligning heterogenous data across technologies, modalities and timepoints, enabling spatiotemporal reconstruction of complex developmental processes.
- Chen-Rui Xia
- , Zhi-Jie Cao
- & Ge Gao
-
Article
| Open Access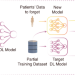 A unified method to revoke the private data of patients in intelligent healthcare with audit to forget
A unified method to revoke the private data of patients in intelligent healthcare with audit to forget
Revoking personal private data is one of the basic human rights. Here, the authors show AFS, a unified method to revoke patients’ private data from pre-trained deep learning models.
- Juexiao Zhou
- , Haoyang Li
- & Xin Gao
-
Article
| Open Access Epitranscriptomic subtyping, visualization, and denoising by global motif visualization
Epitranscriptomic subtyping, visualization, and denoising by global motif visualization
The current available tools lack the ability to accurately classify and visually represent epitranscriptomic profiling data. Here, the authors provide a framework that offers a general solution for the visualization and interpretation of such data.
- Jianheng Liu
- , Tao Huang
- & Rui Zhang
-
Article
| Open Access Mining multi-center heterogeneous medical data with distributed synthetic learning
Mining multi-center heterogeneous medical data with distributed synthetic learning
Here the authors present Distributed Synthetic Learning, a system that addresses data privacy, isolated data islands, and heterogeneity concerns in healthcare analytics by learning to generate state-of-the-art synthetic data for downstream tasks.
- Qi Chang
- , Zhennan Yan
- & Dimitris N. Metaxas
-
Article
| Open Access CRUSTY: a versatile web platform for the rapid analysis and visualization of high-dimensional flow cytometry data
CRUSTY: a versatile web platform for the rapid analysis and visualization of high-dimensional flow cytometry data
CRUSTY is an interactive webtool for flow cytometry data analysis, offering popular algorithms and visualizations, and generating publication-quality figures in minutes. It enables users without bioinformatics expertize to mine complex datasets, supports real-time exploration, and is freely available online.
- Simone Puccio
- , Giorgio Grillo
- & Enrico Lugli
-
Article
| Open Access Data-mining unveils structure–property–activity correlation of viral infectivity enhancing self-assembling peptides
Data-mining unveils structure–property–activity correlation of viral infectivity enhancing self-assembling peptides
Certain peptides can boost viral infectivity. However, the requirements for their activity remain unclear. Here, the authors demonstrate that peptides are efficient viral enhancers if they form hydrophobic β-sheet-rich, positively charged μm-sized aggregates.
- Kübra Kaygisiz
- , Lena Rauch-Wirth
- & Tanja Weil
-
Article
| Open Access DECIMER.ai: an open platform for automated optical chemical structure identification, segmentation and recognition in scientific publications
DECIMER.ai: an open platform for automated optical chemical structure identification, segmentation and recognition in scientific publications
Chemical structures are typically published as nonmachine-readable images in scientific literature. Here, the authors present DECIMER.ai, an open platform for translating chemical structures in publications into machine-readable representations.
- Kohulan Rajan
- , Henning Otto Brinkhaus
- & Christoph Steinbeck
-
Article
| Open Access Cell-type-specific co-expression inference from single cell RNA-sequencing data
Cell-type-specific co-expression inference from single cell RNA-sequencing data
Inferring co-expressions with scRNA-seq data is challenging, and existing methods suffer from inflated false positives and biases. Here, the authors proposed CS-CORE, which yields unbiased estimates and identifies co-expressions that are more reproducible and biologically relevant for scRNA-seq data.
- Chang Su
- , Zichun Xu
- & Jingfei Zhang
-
Article
| Open Access Cellular state landscape and herpes simplex virus type 1 infection progression are connected
Cellular state landscape and herpes simplex virus type 1 infection progression are connected
The heterogeneity of single cell responses during infection have been reported to influence disease outcome. Here, Pietilä et al characterize cellular heterogeneity during Herpes Simplex Virus 1 infection using a multimodal approach that resolves gene expression, proteomic and spatial details at the single cell level.
- Maija K. Pietilä
- , Jana J. Bachmann
- & Cornel Fraefel
-
Article
| Open Access Multi-omics analysis of human mesenchymal stem cells shows cell aging that alters immunomodulatory activity through the downregulation of PD-L1
Multi-omics analysis of human mesenchymal stem cells shows cell aging that alters immunomodulatory activity through the downregulation of PD-L1
Mesenchymal stem cells (MSC) are used for immunosuppressive therapy and a uniform source or heterogeneity characterisation is needed. Here the authors use multi-omics to compare human MSC from different sources and ages of donors and show differences in gene expression and immunosuppressive function.
- Yuchen Gao
- , Ying Chi
- & Xiaomin Zhang
-
Article
| Open Access Detecting diagnostic features in MS/MS spectra of post-translationally modified peptides
Detecting diagnostic features in MS/MS spectra of post-translationally modified peptides
Protein modifications increase the complexity of data analysis in mass spectrometry-based proteomics, which may impair the comprehensive mapping of modification sites. Here, the authors develop an algorithm to extract diagnostic fragmentation patterns to improve modified peptide recovery and localization.
- Daniel J. Geiszler
- , Daniel A. Polasky
- & Alexey I. Nesvizhskii
-
Article
| Open Access CAJAL enables analysis and integration of single-cell morphological data using metric geometry
CAJAL enables analysis and integration of single-cell morphological data using metric geometry
Cell morphology is one of the most described phenotypes in biology, yet systematic quantification and classification of morphology remains limited. Here, the authors present a computational approach for cell morphometry and multi-modal analysis based on concepts from metric geometry.
- Kiya W. Govek
- , Patrick Nicodemus
- & Pablo G. Camara
-
Article
| Open Access Identification of CircRNA signature associated with tumor immune infiltration to predict therapeutic efficacy of immunotherapy
Identification of CircRNA signature associated with tumor immune infiltration to predict therapeutic efficacy of immunotherapy
Circular RNAs are known to be linked to cancer regulation. Here, the authors identify a circular RNA signature associated with immune checkpoint response in melanoma.
- Yu Dong
- , Qian Gao
- & Youqiong Ye
-
Article
| Open Access DeepFLR facilitates false localization rate control in phosphoproteomics
DeepFLR facilitates false localization rate control in phosphoproteomics
Protein phosphorylation is a critical modification in many cellular processes. Here, the authors present DeepFLR, a deep learning-based framework to accurately predict phosphopeptide tandem mass spectra and effectively control false localization rates in phosphoproteomics.
- Yu Zong
- , Yuxin Wang
- & Liang Qiao
-
Article
| Open Access MS2Query: reliable and scalable MS2 mass spectra-based analogue search
MS2Query: reliable and scalable MS2 mass spectra-based analogue search
The authors develop a machine learning approach to find structurally related chemicals in mass spectral libraries. Their method boosts the annotation rate and aids in assessing novelty in metabolomics datasets.
- Niek F. de Jonge
- , Joris J. R. Louwen
- & Justin J. J. van der Hooft
-
Article
| Open Access Lacking mechanistic disease definitions and corresponding association data hamper progress in network medicine and beyond
Lacking mechanistic disease definitions and corresponding association data hamper progress in network medicine and beyond
Large-scale disease-association data are widely used for pathomechanism mining, even if disease definitions used for annotation are mostly phenotype-based. Here, the authors show that this bias can lead to a blurred view on disease mechanisms, highlighting the need for close-up studies based on molecular data for well-characterized patient cohorts.
- Sepideh Sadegh
- , James Skelton
- & David B. Blumenthal
-
Article
| Open Access Multi-modal quantification of pathway activity with MAYA
Multi-modal quantification of pathway activity with MAYA
Pathways can be activated through various signaling cascades depending on cell type. Here, the authors introduce MAYA, a computational method that can detect and score multiple modes of activation for each pathway, improving the granularity of pathway analysis for single-cell datasets.
- Yuna Landais
- & Céline Vallot
-
Article
| Open Access Virtual elastography ultrasound via generative adversarial network for breast cancer diagnosis
Virtual elastography ultrasound via generative adversarial network for breast cancer diagnosis
The current use of elastography ultrasound faces challenges, including vulnerability to subjective manipulation, echo signal attenuation, unknown risks of elastic pressure and high imaging hardware cost. Here, the author shows a virtual elastography to empower low-end ultrasound devices with state-of-art elastography function.
- Zhao Yao
- , Ting Luo
- & JianQiao Zhou
-
Article
| Open Access Integrative proteomic characterization of adenocarcinoma of esophagogastric junction
Integrative proteomic characterization of adenocarcinoma of esophagogastric junction
The molecular subtypes of adenocarcinoma of the esophagogastric junction (AEG) remain to be identified. Here, the authors perform proteogenomic characterisation of AEG tumours with paired normal adjacent tissues and suggest three proteomic subtypes and potential druggable targets.
- Shengli Li
- , Li Yuan
- & Xiang-Dong Cheng
-
Article
| Open Access Multilingual translation for zero-shot biomedical classification using BioTranslator
Multilingual translation for zero-shot biomedical classification using BioTranslator
Here, the authors develop the cross-modal translation method BioTranslator to translate the textual description to non-text biological data. This approach frees scientists from limiting their analysis within predefined controlled vocabularies.
- Hanwen Xu
- , Addie Woicik
- & Sheng Wang
-
Article
| Open Access Reanalysis of ribosome profiling datasets reveals a function of rocaglamide A in perturbing the dynamics of translation elongation via eIF4A
Reanalysis of ribosome profiling datasets reveals a function of rocaglamide A in perturbing the dynamics of translation elongation via eIF4A
The compound Rocaglamide A (RocA) is known for repressing translation initiation. Here the authors identify a dual mode of action for RocA in blocking translation initiation and elongation via eIF4A using previous datasets and new analyses.
- Fajin Li
- , Jianhuo Fang
- & Xuerui Yang
-
Article
| Open Access Genomic disparities between cancers in adolescent and young adults and in older adults
Genomic disparities between cancers in adolescent and young adults and in older adults
The biological underpinnings underlying the increased mortality and morbidity in adolescents and young adults (AYA) remains poorly understood. Here, the authors investigate the clinical and genomic disparities in AYA and older adults in a cohort of more than 100,000 cancer patients.
- Xiaojing Wang
- , Anne-Marie Langevin
- & Siyuan Zheng
-
Article
| Open Access CLIMB: High-dimensional association detection in large scale genomic data
CLIMB: High-dimensional association detection in large scale genomic data
Comparisons among experimental results with large amounts of data can be more precise and meaningful when done across multiple different conditions simultaneously. Koch et al. introduce a method, called CLIMB, that does this, and captures interpretable and biologically meaningful information.
- Hillary Koch
- , Cheryl A. Keller
- & Qunhua Li
-
Article
| Open Access Systematic characterization of cancer transcriptome at transcript resolution
Systematic characterization of cancer transcriptome at transcript resolution
Modification of transcribed mRNAs enables regulation of transcription but its extent in cancer cells is incompletely understood. Here, the authors analyse transcript assembly in over 1000 cancer cell lines and find unannotated transcripts are common, and are associated with drug sensitivity.
- Wei Hu
- , Yangjun Wu
- & Shengli Li
-
Article
| Open Access Systematic tissue annotations of genomics samples by modeling unstructured metadata
Systematic tissue annotations of genomics samples by modeling unstructured metadata
The 1+ million publicly-available human –omics samples currently remain acutely underused. Here the authors present an approach combining natural language processing and machine learning to infer the source tissue of public genomics samples based on their plain text descriptions, making these samples easy to discover and reuse.
- Nathaniel T. Hawkins
- , Marc Maldaver
- & Arjun Krishnan
-
Article
| Open Access Multi-omics characterization of autophagy-related molecular features for therapeutic targeting of autophagy
Multi-omics characterization of autophagy-related molecular features for therapeutic targeting of autophagy
Autophagy has been typically associated with resistance to cancer therapy, and autophagy inhibitors have been explored in cancer. Here, the authors investigate autophagy signatures and their association with drug response in cancer, and find that autophagy induction can actually sensitise cancer cells to therapy.
- Mei Luo
- , Lin Ye
- & Leng Han
-
Article
| Open Access Alignment of single-cell trajectory trees with CAPITAL
Alignment of single-cell trajectory trees with CAPITAL
Global alignment of complex cell state trajectories between single-cell datasets remains challenging. Here, the authors present a computational method called CAPITAL to compare branching trajectories, and demonstrate that this method achieves accurate and robust alignments.
- Reiichi Sugihara
- , Yuki Kato
- & Yukio Kawahara
-
Article
| Open Access Clustering by measuring local direction centrality for data with heterogeneous density and weak connectivity
Clustering by measuring local direction centrality for data with heterogeneous density and weak connectivity
Clustering is a powerful machine learning method for discovering similar patterns according to the proximity of elements in feature space. Here the authors propose a local direction centrality clustering algorithm that copes with heterogeneous density and weak connectivity issues.
- Dehua Peng
- , Zhipeng Gui
- & Huayi Wu
-
Article
| Open Access Connecting omics signatures and revealing biological mechanisms with iLINCS
Connecting omics signatures and revealing biological mechanisms with iLINCS
There are only a few platforms that integrate multiple omics data types, bioinformatics tools, and interfaces for integrative analyses and visualization that do not require programming skills. Here the authors present an integrative web-based platform for analysis of omics data and signatures of cellular perturbations.
- Marcin Pilarczyk
- , Mehdi Fazel-Najafabadi
- & Mario Medvedovic
-
Article
| Open Access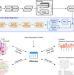 GenomicSuperSignature facilitates interpretation of RNA-seq experiments through robust, efficient comparison to public databases
GenomicSuperSignature facilitates interpretation of RNA-seq experiments through robust, efficient comparison to public databases
Many transcriptomic profiles have been deposited in public archives but are underused for the interpretation of experiments. Here the authors report GenomicSuperSignature for interpreting new transcriptomic datasets through comparison to public archives, without high-performance computing requirements.
- Sehyun Oh
- , Ludwig Geistlinger
- & Sean Davis
-
Article
| Open Access Global stable-isotope tracing metabolomics reveals system-wide metabolic alternations in aging Drosophila
Global stable-isotope tracing metabolomics reveals system-wide metabolic alternations in aging Drosophila
Stable-isotope tracing allows quantifying metabolic activity by measuring isotopically labeled metabolites, but its metabolome coverage has been limited. Here, the authors develop a global isotope tracing approach with metabolome-wide coverage and use it to characterize metabolic activities in aging Drosophila.
- Ruohong Wang
- , Yandong Yin
- & Zheng-Jiang Zhu
-
Article
| Open Access Exploring the cellular landscape of circular RNAs using full-length single-cell RNA sequencing
Exploring the cellular landscape of circular RNAs using full-length single-cell RNA sequencing
Studies of circular RNAs have often been limited to the tissue or organism level. Here, authors investigate the comprehensive expression landscape of circRNAs in human and mouse at single-cell resolution, revealing highly specific and dynamic changes of circRNAs during multiple biological processes.
- Wanying Wu
- , Jinyang Zhang
- & Fangqing Zhao
-
Article
| Open Access Short- and long-read metagenomics expand individualized structural variations in gut microbiomes
Short- and long-read metagenomics expand individualized structural variations in gut microbiomes
Here, Wang and colleagues combine short and long sequencing reads to characterize structural variations, prophage and CRISPR spacer elements in human gut microbiomes, and reveal functional differences at a finer level of bacterial strains.
- Liang Chen
- , Na Zhao
- & Jun Wang
-
Article
| Open Access Mutational signatures are markers of drug sensitivity of cancer cells
Mutational signatures are markers of drug sensitivity of cancer cells
Mutational signatures can reveal the impact of mutagenic processes in cancer, including exposure to therapy. Here, the authors develop an approach that can accurately predict drug responses in cancer using mutational signatures while simultaneously correcting for germline variants with an ancestry matching procedure.
- Jurica Levatić
- , Marina Salvadores
- & Fran Supek
-
Article
| Open Access Persistent COVID-19 symptoms in a community study of 606,434 people in England
Persistent COVID-19 symptoms in a community study of 606,434 people in England
This study characterises Long COVID using data from the REACT-2 community-based study in England. It estimates that 38% (in autumn/winter 2020/21) and 22% (in spring 2021) of people reported at least one symptom 12 weeks after symptom onset; identifies risk factors for persistent symptoms; and finds evidence of symptom clustering.
- Matthew Whitaker
- , Joshua Elliott
- & Paul Elliott
 scButterfly: a versatile single-cell cross-modality translation method via dual-aligned variational autoencoders
scButterfly: a versatile single-cell cross-modality translation method via dual-aligned variational autoencoders