
Featured
-
-
Article
| Open Access Accelerated discovery of superoxide-dismutase nanozymes via high-throughput computational screening
Accelerated discovery of superoxide-dismutase nanozymes via high-throughput computational screening
A general predicting theory for superoxide-dismutase mimicking nanomaterials is currently lacking. The present manuscript reports a density functional theory study on the superoxides dismutase-like activity of nanomaterials based on their electronic band structures and surface adsorption energies.
- Zhenzhen Wang
- , Jiangjiexing Wu
- & Yuliang Zhao
-
Article
| Open Access Proofreading experimentally assigned stereochemistry through Q2MM predictions in Pd-catalyzed allylic aminations
Proofreading experimentally assigned stereochemistry through Q2MM predictions in Pd-catalyzed allylic aminations
A predictive model has been created for a stereoselective palladium-catalysed allylic amination reaction. Derived only from quantum chemical data, the method is accurate enough to reveal multiple erroneous assignments in literature experiments.
- Jessica Wahlers
- , Jèssica Margalef
- & Per-Ola Norrby
-
Article
| Open Access Cross-property deep transfer learning framework for enhanced predictive analytics on small materials data
Cross-property deep transfer learning framework for enhanced predictive analytics on small materials data
Artificial intelligence and machine learning can greatly enhance materials property prediction and discovery. Here the authors propose cross-property transfer learning to build accurate models for dozens of properties with limited data availability.
- Vishu Gupta
- , Kamal Choudhary
- & Ankit Agrawal
-
Article
| Open Access Monovalent lanthanide(I) in borozene complexes
Monovalent lanthanide(I) in borozene complexes
The most common oxidation state for lanthanides is +3. Here the authors use photoelectron spectroscopy and theoretical calculations to study half-sandwich complexes where a lanthanide center in the oxidation state +1 is bound to an aromatic wheel-like B82- ligand.
- Wan-Lu Li
- , Teng-Teng Chen
- & Lai-Sheng Wang
-
Article
| Open Access Spontaneous dynamical disordering of borophenes in MgB2 and related metal borides
Spontaneous dynamical disordering of borophenes in MgB2 and related metal borides
Layered boron compounds attract enormous interest in applications. This work reports first-principles calculations coupled with global optimization to show that the outer boron surface in MgB2 nanosheets undergo disordering and clustering, which is experimentally confirmed in synthesized MgB2 nanosheets.
- Sichi Li
- , Harini Gunda
- & Brandon C. Wood
-
Article
| Open Access Robust recognition and exploratory analysis of crystal structures via Bayesian deep learning
Robust recognition and exploratory analysis of crystal structures via Bayesian deep learning
The present manuscript reports a Bayesian deep-learning approach for the automatic, robust classification of polycrystalline systems of both synthetic and experimental origin. The unsupervised analysis of the internal neural-network representations reveals physically understandable patterns.
- Andreas Leitherer
- , Angelo Ziletti
- & Luca M. Ghiringhelli
-
Article
| Open Access Towards complete assignment of the infrared spectrum of the protonated water cluster H+(H2O)21
Towards complete assignment of the infrared spectrum of the protonated water cluster H+(H2O)21
Protonated water species have been the subject of numerous experimental and computational studies. Here the authors provide a nearly complete assignment of the experimental IR spectrum of the H+(H2O)21 water cluster based on high-level wavefunction theory and anharmonic vibrational quasi-degenerate perturbation theory.
- Jinfeng Liu
- , Jinrong Yang
- & Xiao He
-
Article
| Open Access Coupling complementary strategy to flexible graph neural network for quick discovery of coformer in diverse co-crystal materials
Coupling complementary strategy to flexible graph neural network for quick discovery of coformer in diverse co-crystal materials
Experimental determination of new cocrystals remains challenging due to the need of a systematic screening with a large range of coformers. Here the authors develop a flexible deep learning framework based on graph neural network demonstrated to quickly predict the formation of co-crystals.
- Yuanyuan Jiang
- , Zongwei Yang
- & Xuemei Pu
-
Article
| Open Access Two-dimensional monolayer salt nanostructures can spontaneously aggregate rather than dissolve in dilute aqueous solutions
Two-dimensional monolayer salt nanostructures can spontaneously aggregate rather than dissolve in dilute aqueous solutions
Aqueous solutions under nanoscale confinement exhibit interesting physicochemical properties. This work reports evidence on the spontaneous formation of two-dimensional alkali chloride crystalline/non-crystalline nanostructures in dilute aqueous solution under nanoscale confinement by computer simulations.
- Wenhui Zhao
- , Yunxiang Sun
- & Xiao Cheng Zeng
-
Article
| Open Access Real-time dynamics and structures of supported subnanometer catalysts via multiscale simulations
Real-time dynamics and structures of supported subnanometer catalysts via multiscale simulations
Understanding the catalysts’ structure evolution under working conditions is challenging. Here the authors use a multiscale simulation approach and machine learning to study the structures and nucleation of CeO2-supported Pd clusters and single atoms at various catalyst loadings, temperatures, and exposures to CO.
- Yifan Wang
- , Jake Kalscheur
- & Dionisios G. Vlachos
-
Article
| Open Access Relief of excited-state antiaromaticity enables the smallest red emitter
Relief of excited-state antiaromaticity enables the smallest red emitter
Commonly, large π-conjugated systems facilitate low-energy electronic transitions. Here, the authors demonstrate that the relief of excited-state antiaromaticity of the benzene core leads to large Stokes shifts, and allows the construction of emitters covering the entire visible spectrum without the need of extending π-conjugation.
- Heechan Kim
- , Woojin Park
- & Dongwhan Lee
-
Article
| Open Access Facile generation of bridged medium-sized polycyclic systems by rhodium-catalysed intramolecular (3+2) dipolar cycloadditions
Facile generation of bridged medium-sized polycyclic systems by rhodium-catalysed intramolecular (3+2) dipolar cycloadditions
The bridged medium-sized ring bicyclo[m.n.2] family of natural products are commonly found but difficult to synthesize efficiently. Here the authors present a cascade reaction to form the carbon skeleton, via a [3+2] cycloaddition of a captured azavinyl carbene intermediate.
- Bao-Long Hou
- , Jonathan J. Wong
- & Chuang-Chuang Li
-
Article
| Open Access Controllable CO2 electrocatalytic reduction via ferroelectric switching on single atom anchored In2Se3 monolayer
Controllable CO2 electrocatalytic reduction via ferroelectric switching on single atom anchored In2Se3 monolayer
Electroreduction of CO2 into chemical fuels holds promise for mitigating environmental pollution and energy crisis. This work presents a distinct design of ferroelectric catalysts with high catalytic activity and selectivity for efficient and controllable electrochemical CO2 reduction reaction.
- Lin Ju
- , Xin Tan
- & Liangzhi Kou
-
Article
| Open Access Differentiable sampling of molecular geometries with uncertainty-based adversarial attacks
Differentiable sampling of molecular geometries with uncertainty-based adversarial attacks
Neural Networks are known to perform poorly outside of their training domain. Here the authors propose an inverse sampling strategy to train neural network potentials enabling to drive atomistic systems towards high-likelihood and high-uncertainty configurations without the need for molecular dynamics simulations.
- Daniel Schwalbe-Koda
- , Aik Rui Tan
- & Rafael Gómez-Bombarelli
-
Article
| Open Access Teaching a neural network to attach and detach electrons from molecules
Teaching a neural network to attach and detach electrons from molecules
Quantum mechanical calculations of molecular ionized states are computationally quite expensive. This work reports a successful extension of a previous deep-neural networks approach towards transferable neural-network models for predicting multiple properties of open shell anions and cations.
- Roman Zubatyuk
- , Justin S. Smith
- & Olexandr Isayev
-
Article
| Open Access Real space electron delocalization, resonance, and aromaticity in chemistry
Real space electron delocalization, resonance, and aromaticity in chemistry
The concept of delocalization, resonance and aromaticity are commonly discussed within electronic structure frameworks relying on specific wave function expansions. Here the authors propose a redefinition of these concepts from first-principles by investigating saddle points of the all-electron probability density.
- Leonard Reuter
- & Arne Lüchow
-
Article
| Open Access Machine learning based energy-free structure predictions of molecules, transition states, and solids
Machine learning based energy-free structure predictions of molecules, transition states, and solids
Accurate computational prediction of atomistic structure with traditional methods is challenging. The authors report a kernel-based machine learning model capable of reconstructing 3D atomic coordinates from predicted interatomic distances across a variety of system classes.
- Dominik Lemm
- , Guido Falk von Rudorff
- & O. Anatole von Lilienfeld
-
Matters Arising
| Open Access A double bond with weak σ- and strong π-interactions is still a double bond
A double bond with weak σ- and strong π-interactions is still a double bond
- Cina Foroutan-Nejad
-
Article
| Open Access High-pressure reversibility in a plastically flexible coordination polymer crystal
High-pressure reversibility in a plastically flexible coordination polymer crystal
Mechanically flexible single crystals are promising materials for advanced technological applications. Here, the authors study the high pressure response of a plastically flexible coordination polymer and provide indication of an overall disparate mechanical response of bulk flexibility and quasi-hydrostatic compression within the same crystal lattice.
- Xiaojiao Liu
- , Adam A. L. Michalchuk
- & Colin R. Pulham
-
Article
| Open Access Improved prediction of solvation free energies by machine-learning polarizable continuum solvation model
Improved prediction of solvation free energies by machine-learning polarizable continuum solvation model
Accurate theoretical evaluation of solvation free energy is challenging. Here the authors introduce a machine-learning based polarizable continuum solvation approach to improve the accuracy of widely accepted continuum solvation models by almost one order of magnitude without additional computational costs.
- Amin Alibakhshi
- & Bernd Hartke
-
Article
| Open Access Algebraic graph-assisted bidirectional transformers for molecular property prediction
Algebraic graph-assisted bidirectional transformers for molecular property prediction
Despite considerable efforts, quantitative prediction of various molecular properties remains a challenge. Here, the authors propose an algebraic graph-assisted bidirectional transformer, which can incorporate massive unlabeled molecular data into molecular representations via a self-supervised learning strategy and assisted with 3D stereochemical information from graphs.
- Dong Chen
- , Kaifu Gao
- & Feng Pan
-
Article
| Open Access Masked graph modeling for molecule generation
Masked graph modeling for molecule generation
Generating new sensible molecular structures is a key problem in computer aided drug discovery. Here the authors propose a graph-based molecular generative model that outperforms previously proposed graph-based generative models of molecules and performs comparably to several SMILES-based models.
- Omar Mahmood
- , Elman Mansimov
- & Kyunghyun Cho
-
Article
| Open Access Magnetically induced currents and aromaticity in ligand-stabilized Au and AuPt superatoms
Magnetically induced currents and aromaticity in ligand-stabilized Au and AuPt superatoms
Efficient methods to calculate magnetically induced currents in metallic nanostructures are currently lacking. Here, the authors propose a theoretical method to compute and analyze magnetically induced currents in nanostructures validated for experimentally synthesized gold-based, hydrogen-containing ligand-protected clusters.
- Omar López-Estrada
- , Bernardo Zuniga-Gutierrez
- & Hannu Häkkinen
-
Article
| Open Access Active discovery of organic semiconductors
Active discovery of organic semiconductors
Existing methods for organic semiconductor computational screening are limited by the computational demand of the process, leading to the identification of non-optimal material candidates. Here, the authors report machine learning method to guide the discovery of organic semiconductors.
- Christian Kunkel
- , Johannes T. Margraf
- & Karsten Reuter
-
Comment
| Open Access Prospects and challenges for computer simulations of monolayer-protected metal clusters
Prospects and challenges for computer simulations of monolayer-protected metal clusters
Precise knowledge of chemical composition and atomic structure of functional nanosized systems, such as metal clusters stabilized by an organic molecular layer, allows for detailed computational work to investigate structure-property relations. Here, we discuss selected recent examples of computational work that has advanced understanding of how these clusters work in catalysis, how they interact with biological systems, and how they can make self-assembled, macroscopic materials. A growing challenge is to develop effective new simulation methods that take into account the cluster-environment interactions. These new hybrid methods are likely to contain components from electronic structure theory combined with machine learning algorithms for accelerated evaluations of atom-atom interactions.
- Sami Malola
- & Hannu Häkkinen
-
Article
| Open Access Single-atom alloy catalysts designed by first-principles calculations and artificial intelligence
Single-atom alloy catalysts designed by first-principles calculations and artificial intelligence
Single-atom metal alloys attract considerable interest as alternative metal hydrogenation catalysts. Here the authors combine first-principles calculations with compressed-sensing data-analytics approaches to develop stability and activity’s descriptors for screening single atom alloy catalysts.
- Zhong-Kang Han
- , Debalaya Sarker
- & Sergey V. Levchenko
-
Article
| Open Access Quantitative interpretation explains machine learning models for chemical reaction prediction and uncovers bias
Quantitative interpretation explains machine learning models for chemical reaction prediction and uncovers bias
Machine learning algorithms offer new possibilities for automating reaction procedures. The present paper investigates automated reaction’s prediction with Molecular Transformer, the state-of-the-art model for reaction prediction, proposing a new debiased dataset for a realistic assessment of the model’s performance.
- Dávid Péter Kovács
- , William McCorkindale
- & Alpha A. Lee
-
Article
| Open Access The atomistic details of the ice recrystallisation inhibition activity of PVA
The atomistic details of the ice recrystallisation inhibition activity of PVA
Understanding ice re-crystallization is key to improve the current cryopreservation technologies. Here, the authors bring together experiments and simulations to unravel the atomistic details of the ice re-crystallization inhibition (IRI) activity of poly(vinyl)alcohol—the most potent biomimetic IRI agent.
- Fabienne Bachtiger
- , Thomas R. Congdon
- & Gabriele C. Sosso
-
Article
| Open Access Automated discovery of a robust interatomic potential for aluminum
Automated discovery of a robust interatomic potential for aluminum
The accuracy of a machine-learned potential is limited by the quality and diversity of the training dataset. Here the authors propose an active learning approach to automatically construct general purpose machine-learning potentials here demonstrated for the aluminum case.
- Justin S. Smith
- , Benjamin Nebgen
- & Kipton Barros
-
Article
| Open Access Simulating the ghost: quantum dynamics of the solvated electron
Simulating the ghost: quantum dynamics of the solvated electron
The nature of the bulk hydrated electron has been a challenge for both experiment and theory. Here the authors use a machine-learning model trained on MP2 data to achieve an accurate determination of the structure, diffusion mechanisms, and vibrational spectroscopy of the solvated electron.
- Jinggang Lan
- , Venkat Kapil
- & Vladimir V. Rybkin
-
Article
| Open Access Quantum-mechanical exploration of the phase diagram of water
Quantum-mechanical exploration of the phase diagram of water
Complex interatomic interactions and diverse structures make computing the phase diagram of water very challenging. Here, a combination of machine learning and advanced free-energy methods at three levels of hybrid DFT enables the prediction of the phase diagram in close agreement with experiment.
- Aleks Reinhardt
- & Bingqing Cheng
-
Article
| Open Access Dynamical strengthening of covalent and non-covalent molecular interactions by nuclear quantum effects at finite temperature
Dynamical strengthening of covalent and non-covalent molecular interactions by nuclear quantum effects at finite temperature
The inclusion of nuclear quantum effects (NQE) in atomistic simulations of chemical systems is of key importance. Here the authors use machine learned force fields trained on coupled cluster reference data to show the dynamical strengthening of covalent and non-covalent molecular interactions induced by NQE.
- Huziel E. Sauceda
- , Valentin Vassilev-Galindo
- & Alexandre Tkatchenko
-
Article
| Open Access Pure non-local machine-learned density functional theory for electron correlation
Pure non-local machine-learned density functional theory for electron correlation
Semilocal density functionals, while computationally efficient, do not account for non-local correlation. Here, the authors propose a machine-learning approach to DFT that leads to non-local and transferable functionals applicable to non-covalent, ionic and covalent interactions across system of different sizes.
- Johannes T. Margraf
- & Karsten Reuter
-
Article
| Open Access Coulomb interactions between dipolar quantum fluctuations in van der Waals bound molecules and materials
Coulomb interactions between dipolar quantum fluctuations in van der Waals bound molecules and materials
High-level methods to describe van der Waals interactions are limited due to their computational cost. This work introduces a new theoretical approach, that extends the dipolar many-body dispersion formalism to higher-order contributions, demonstrated to be applicable to practically-relevant systems and nano-environments.
- Martin Stöhr
- , Mainak Sadhukhan
- & Alexandre Tkatchenko
-
Article
| Open Access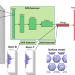 Machine learned features from density of states for accurate adsorption energy prediction
Machine learned features from density of states for accurate adsorption energy prediction
Computational catalysis would strongly benefit from general descriptors applicable for predicting adsorption energetics. Here the authors propose a machine-learning approach for adsorption energy predictions based on learning the relevant descriptors in a surface atom's density of states as part of the training.
- Victor Fung
- , Guoxiang Hu
- & Bobby G. Sumpter
-
Article
| Open Access Key activity descriptors of nickel-iron oxygen evolution electrocatalysts in the presence of alkali metal cations
Key activity descriptors of nickel-iron oxygen evolution electrocatalysts in the presence of alkali metal cations
It is commonly accepted that electrolyte alkali metal cations modify the catalytic activity for oxygen evolution reaction. Here the authors challenge this assumption, showing that the activity is actually affected by a change in the electrolyte pH rather than a specific alkali cation.
- Mikaela Görlin
- , Joakim Halldin Stenlid
- & Oscar Diaz-Morales
-
Article
| Open Access Machine learning with physicochemical relationships: solubility prediction in organic solvents and water
Machine learning with physicochemical relationships: solubility prediction in organic solvents and water
Accurate prediction of solubility represents a challenge for traditional computational approaches due to the complex nature of phenomena involved. Here the authors report a successful approach to solubility prediction in organic solvents and water using combination of machine learning and computational chemistry.
- Samuel Boobier
- , David R. J. Hose
- & Bao N. Nguyen
-
Article
| Open Access Liquid water contains the building blocks of diverse ice phases
Liquid water contains the building blocks of diverse ice phases
Molecular understanding of water is challenging due to the structural complexity of liquid water and the large number of ice phases. Here the authors use a machine-learning potential trained on liquid water to demonstrate the structural similarity of liquid water and that of 54 real and hypothetical ice phases.
- Bartomeu Monserrat
- , Jan Gerit Brandenburg
- & Bingqing Cheng
-
Article
| Open Access Complex reaction processes in combustion unraveled by neural network-based molecular dynamics simulation
Complex reaction processes in combustion unraveled by neural network-based molecular dynamics simulation
Gaining insights into combustion processes is challenging due to the complex reactions involved. The present work proposes a neural network potential model trained to ab initio data that enables to simulate the combustion of methane by predicting reactants, products and reaction intermediates.
- Jinzhe Zeng
- , Liqun Cao
- & John Z. H. Zhang
-
Article
| Open Access State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis
State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis
Development of algorithms to predict reactant and reagents given a target molecule is key to accelerate retrosynthesis approaches. Here the authors demonstrate that applying augmentation techniques to the SMILE representation of target data significantly improves the quality of the reaction predictions.
- Igor V. Tetko
- , Pavel Karpov
- & Guillaume Godin
-
Article
| Open Access Machine learning in chemical reaction space
Machine learning in chemical reaction space
Application of machine-learning approaches to exploring chemical reaction networks is challenging due to need of including open-shell reaction intermediates. Here the authors introduce a density functional theory database of closed and open-shell molecules for machine-learning predictions of reaction energies.
- Sina Stocker
- , Gábor Csányi
- & Johannes T. Margraf
-
Article
| Open Access Infrared spectroscopic study of hydrogen bonding topologies in the smallest ice cube
Infrared spectroscopic study of hydrogen bonding topologies in the smallest ice cube
Spectroscopic studies of water clusters provide insight into the hydrogen bond structure of water and ice. The authors measure infrared spectra of neutral water octamers using a threshold photoionization technique based on a tunable vacuum-UV free electron laser, identifying two cubic isomers in addition to those previously observed.
- Gang Li
- , Yang-Yang Zhang
- & Ling Jiang
-
Article
| Open Access Quantum chemical accuracy from density functional approximations via machine learning
Quantum chemical accuracy from density functional approximations via machine learning
High-level ab initio quantum chemical methods carry a high computational burden, thus limiting their applicability. Here, the authors employ machine learning to generate coupled-cluster energies and forces at chemical accuracy for geometry optimization and molecular dynamics from DFT densities.
- Mihail Bogojeski
- , Leslie Vogt-Maranto
- & Kieron Burke
-
Article
| Open Access Optical gap and fundamental gap of oligoynes and carbyne
Optical gap and fundamental gap of oligoynes and carbyne
Carbyne, a linear sp-hybridized carbon allotrope, is synthetically inaccessible and its properties are extrapolated from those of defined oligomers. Here the authors analyze weak optical bands in two series of oligoynes and reassess the optical and fundamental gap of carbyne to lower values than previously suggested.
- Johannes Zirzlmeier
- , Stephen Schrettl
- & Holger Frauenrath
-
Article
| Open Access Automated extraction of chemical synthesis actions from experimental procedures
Automated extraction of chemical synthesis actions from experimental procedures
Extracting experimental operations for chemical synthesis from procedures reported in prose is a tedious task. Here the authors develop a deep-learning model based on the transformer architecture to translate experimental procedures from the field of organic chemistry into synthesis actions.
- Alain C. Vaucher
- , Federico Zipoli
- & Teodoro Laino
-
Article
| Open Access Representation of molecular structures with persistent homology for machine learning applications in chemistry
Representation of molecular structures with persistent homology for machine learning applications in chemistry
The choice of molecular representations can severely impact the performances of machine-learning methods. Here the authors demonstrate a persistence homology based molecular representation through an active-learning approach for predicting CO2/N2 interaction energies at the density functional theory (DFT) level.
- Jacob Townsend
- , Cassie Putman Micucci
- & Konstantinos D. Vogiatzis
-
Article
| Open Access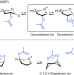 Characterization of glycosyl dioxolenium ions and their role in glycosylation reactions
Characterization of glycosyl dioxolenium ions and their role in glycosylation reactions
Dioxolenium ion intermediates formed from remote positions are hypothesized to direct stereoselective glycosylations. Herein we combine infrared ion spectroscopy, DFT calculations and synthetic work to characterize and study these dioxolenium ions and their role in stereoselective glycosylation reactions.
- Thomas Hansen
- , Hidde Elferink
- & Thomas J. Boltje
-
Article
| Open Access Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost
Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost
Bond dissociation enthalpies are key quantities in determining chemical reactivity, their computations with quantum mechanical methods being highly demanding. Here the authors develop a machine learning approach to calculate accurate dissociation enthalpies for organic molecules with sub-second computational cost.
- Peter C. St. John
- , Yanfei Guan
- & Robert S. Paton
-
Article
| Open Access δ and φ back-donation in AnIV metallacycles
δ and φ back-donation in AnIV metallacycles
Metal-ligand δ and φ interactions, though considered weak, may be necessary for fully describing the electronic and geometric structures of certain compounds. Here, in actinide metallacycles, the authors discover two new types of M-L δ and φ back-bonds that contribute substantially to their unusual chemical behavior.
- Morgan P. Kelley
- , Ivan A. Popov
- & Ping Yang
 Augmenting zero-Kelvin quantum mechanics with machine learning for the prediction of chemical reactions at high temperatures
Augmenting zero-Kelvin quantum mechanics with machine learning for the prediction of chemical reactions at high temperatures