
Featured
-
-
Article
| Open Access Comprehensive identification of internal structure and alternative splicing events in circular RNAs
Comprehensive identification of internal structure and alternative splicing events in circular RNAs
Circular RNAs are increasingly understood to have important biological roles and have several subclasses. Here, the authors develop CIRI-AS to analyse sequencing data, identifying the prevalence of alternative splicing and circular RNA isoforms.
- Yuan Gao
- , Jinfeng Wang
- & Fangqing Zhao
-
Article
| Open Access Mapping RNA–RNA interactome and RNA structure in vivo by MARIO
Mapping RNA–RNA interactome and RNA structure in vivo by MARIO
Current methods for mapping RNA-RNA interactions have to rely on an ‘anchor’ protein or RNA. Here, the authors report the MARIO (Mapping RNA interactome in vivo) technology that can massively reveal RNA-RNA interactions and RNA structure from unperturbed cells.
- Tri C. Nguyen
- , Xiaoyi Cao
- & Sheng Zhong
-
Article
| Open Access Robust estimates of overall immune-repertoire diversity from high-throughput measurements on samples
Robust estimates of overall immune-repertoire diversity from high-throughput measurements on samples
Diversity of an organism’s B- and T-cell repertoires is clinically important, but difficult to estimate due to uncertainty in the number of clones in a sample, sampling bias and experimental noise. Here Kaplinsky and Arnaout present Recon, a method that reconstructs the distribution of the overall repertoire from sample measurements.
- Joseph Kaplinsky
- & Ramy Arnaout
-
Article
| Open Access An organelle-specific protein landscape identifies novel diseases and molecular mechanisms
An organelle-specific protein landscape identifies novel diseases and molecular mechanisms
Mutations in proteins that localize to primary cilia cause devastating diseases, yet the primary cilium is a poorly understood organelle. Here the authors use interaction proteomics to identify a network of human ciliary proteins that provides new insights into several biological processes and diseases.
- Karsten Boldt
- , Jeroen van Reeuwijk
- & Kathy Williamson
-
Article
| Open Access The complete local genotype–phenotype landscape for the alternative splicing of a human exon
The complete local genotype–phenotype landscape for the alternative splicing of a human exon
Genotype–phenotype landscapes are an important characteristic for understanding the evolution of traits. Here the authors construct the local landscape for the alternative splicing of FAS/CD95 exon 6, revealing the regulation of splicing and the evolution of regulatory information between species.
- Philippe Julien
- , Belén Miñana
- & Ben Lehner
-
Article
| Open Access A uniform survey of allele-specific binding and expression over 1000-Genomes-Project individuals
A uniform survey of allele-specific binding and expression over 1000-Genomes-Project individuals
Using variants from the 1000 Genomes Project, RNA-seq and ChIP-seq data from related projects, this study describes a resource and survey of allele-specific binding and gene expression. A catalogue of allelic SNPs and annotation elements is available as an online resource at alleledb.gersteinlab.org.
- Jieming Chen
- , Joel Rozowsky
- & Mark Gerstein
-
Article
| Open Access Characterization of eukaryotic DNA N6-methyladenine by a highly sensitive restriction enzyme-assisted sequencing
Characterization of eukaryotic DNA N6-methyladenine by a highly sensitive restriction enzyme-assisted sequencing
DNA N6-methyladenine is prevalent in prokaryotes, and is recently also detected in eukaryotes such as roundworm and fly. Here, Luo et al. report a DpnI-assisted base resolution method that detects 6mA genome-wide with nanograms of input DNA and lower sequencing depth than the previous restriction enzyme-based approach.
- Guan-Zheng Luo
- , Fang Wang
- & Chuan He
-
Article
| Open Access Fast and sensitive mapping of nanopore sequencing reads with GraphMap
Fast and sensitive mapping of nanopore sequencing reads with GraphMap
Read mapping and alignment tools are critical for many applications based on MinION sequencers. Here, the authors present GraphMap, a mapping algorithm designed to analyze nanopore sequencing reads, that progressively refines candidate alignments to handle potentially high error rates to align long reads.
- Ivan Sović
- , Mile Šikić
- & Niranjan Nagarajan
-
Article
| Open Access Structural hot spots for the solubility of globular proteins
Structural hot spots for the solubility of globular proteins
Mutations in aggregation prone regions of recombinant proteins often improve their solubility, although they might cause negative effects on their structure and function. Here, the authors identify proteins hot spots that can be exploited to optimize solubility without compromising stability.
- Ashok Ganesan
- , Aleksandra Siekierska
- & Joost Schymkowitz
-
Article
| Open Access Probabilistic modelling of chromatin code landscape reveals functional diversity of enhancer-like chromatin states
Probabilistic modelling of chromatin code landscape reveals functional diversity of enhancer-like chromatin states
The chromatin functional state can be derived from the binding patterns of chromatin factors. Here, Zhou and Troyanskaya report a data-driven probabilistic modelling of dependencies between chromatin factors that can divide enhancer-like chromatin states into three functionally distinct groups.
- Jian Zhou
- & Olga G. Troyanskaya
-
Article
| Open Access Network-based in silico drug efficacy screening
Network-based in silico drug efficacy screening
Attempts to predict novel use for existing drugs rarely consider information on the impact on the genes perturbed in a given disease. Here, the authors present a novel network-based drug-disease proximity measure that provides insight on gene specific therapeutic effect of drugs and may facilitate drug repurposing.
- Emre Guney
- , Jörg Menche
- & Albert-László Barábasi
-
Article
| Open Access Iron Age and Anglo-Saxon genomes from East England reveal British migration history
Iron Age and Anglo-Saxon genomes from East England reveal British migration history
This study examines ancient genomes of individuals from the late Iron Age to the middle Anglo-Saxon period in the East of England. Using a newly devised analytic algorithm, the author also estimate the relative ancestry of East English genome derived from Anglo-Saxon migrations and to the rest of Europe.
- Stephan Schiffels
- , Wolfgang Haak
- & Richard Durbin
-
Article
| Open Access Label-free cell cycle analysis for high-throughput imaging flow cytometry
Label-free cell cycle analysis for high-throughput imaging flow cytometry
Imaging flow cytometry enables high-throughput acquisition of fluorescence, brightfield and darkfield images of biological cells. Here, Blasi et al.demonstrate that applying machine learning algorithms on brightfield and darkfield images can detect cellular phenotypes without the need for fluorescent stains, enabling label-free assays.
- Thomas Blasi
- , Holger Hennig
- & Paul Rees
-
Article
| Open Access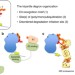 Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin-proteasome system
Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin-proteasome system
Degrons are determinants within proteins that direct programmed degradation by the ubiquitinproteasome system. Here, the authors propose a three-part degron architecture which contains an E3-ligase recognition motif, a ubiquitination site(s), and a disordered site to initiate degradation.
- Mainak Guharoy
- , Pallab Bhowmick
- & Peter Tompa
-
Article
| Open Access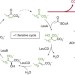 Integrative genomic mining for enzyme function to enable engineering of a non-natural biosynthetic pathway
Integrative genomic mining for enzyme function to enable engineering of a non-natural biosynthetic pathway
The modification of enzymes to generate desired compounds by non-natural pathways is a useful route for the production of commodity chemicals. Here, the authors show two approaches—genome mining and computational enzyme design—to generate higher alcohols from sugar.
- Wai Shun Mak
- , Stephen Tran
- & Justin B. Siegel
-
Article
| Open Access MicroRNA–mRNA interactions underlying colorectal cancer molecular subtypes
MicroRNA–mRNA interactions underlying colorectal cancer molecular subtypes
Colorectal cancer subtypes can be distinguished by their different biological and molecular properties. Here the authors present microRNA Master Regulator Analysis, a tool to identify microRNAs driving subtype-specific gene expression and cancer variation.
- Laura Cantini
- , Claudio Isella
- & Enzo Medico
-
Article
| Open Access A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma
A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma
Using molecular markers is a useful way to predict the prognosis of cancer patients. Here, Wei et al.describe a five gene methylation signature that can predict the prognosis of renal clear cell cancer and validate its use in multiple patient cohorts.
- Jin-Huan Wei
- , Ahmed Haddad
- & Jun-Hang Luo
-
Article
| Open Access ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo
ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo
In chromatin endogenous cleavage (ChEC), micrococcal nuclease (MNase) is fused to a protein of interest and its cleavage is thus targeted to specific genomic loci in vivo. Here, the authors show that time-resolved ChEC-seq (high-throughput sequencing after ChEC) can detect DNA shape patterns regardless of motif strength.
- Gabriel E. Zentner
- , Sivakanthan Kasinathan
- & Steven Henikoff
-
Article
| Open Access The transcriptional landscape of age in human peripheral blood
The transcriptional landscape of age in human peripheral blood
Ageing increases the risk of many diseases. Here the authors compare blood cell transcriptomes of over 14,000 individuals and identify a set of about 1,500 genes that are differently expressed with age, shedding light on transcriptional programs linked to the ageing process and age-associated diseases.
- Marjolein J. Peters
- , Roby Joehanes
- & Andrew D. Johnson
-
Article
| Open Access Harnessing the landscape of microbial culture media to predict new organism–media pairings
Harnessing the landscape of microbial culture media to predict new organism–media pairings
Culturing new microorganisms requires a great deal of experience, and trial and error. Here, the authors build a database of >3,300 culturing media recipes and >18,000 microbial species that allows the prediction of appropriate media recipes for the growth of new microbes based on their 16S rDNA sequences.
- Matthew A. Oberhardt
- , Raphy Zarecki
- & Eytan Ruppin
-
Article
| Open Access Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera
Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera
Lactobacillus is a lactic acid bacteria and has a wide range of application from use in probiotic food production to biotherapeutics. Here, the authors sequence and compare the genomes of 213 different Lactobacillusstrains and related genera, and provide new insight into phylogenomic organization and adaptive immunity elements in this bacteria family.
- Zhihong Sun
- , Hugh M. B. Harris
- & Paul W. O’Toole
-
Article
| Open Access Combining genomic and network characteristics for extended capability in predicting synergistic drugs for cancer
Combining genomic and network characteristics for extended capability in predicting synergistic drugs for cancer
Predicting combinations of chemotherapeutic drugs that act synergistically is challenging. Here the authors take a computational approach to predict synergistic pairs, validate novel pairs using several cancer cell lines, and assess toxicity in a zebrafish xenograft model.
- Yi Sun
- , Zhen Sheng
- & Zhiwei Cao
-
Article
| Open Access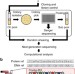 De novo assembly and next-generation sequencing to analyse full-length gene variants from codon-barcoded libraries
De novo assembly and next-generation sequencing to analyse full-length gene variants from codon-barcoded libraries
This paper described a new and efficient method for de novoassembly of multiple DNA sequence information from mutagenized clone libraries. Using codon-barcoded libraries and calling the method JigsawSeq, the authors overcome limitations of short-read sequencing assembly from next-generation sequencing.
- Namjin Cho
- , Byungjin Hwang
- & Duhee Bang
-
Article
| Open Access RNA regulatory networks diversified through curvature of the PUF protein scaffold
RNA regulatory networks diversified through curvature of the PUF protein scaffold
Members of the PUF family of RNA-binding proteins bind multiple mRNAs in vivo. Here the authors show that the S. cerevisiaePuf5p binds targets of varying lengths that correlate with biological functions. The RNA-binding sites adopt different structures to adapt to a fixed protein scaffold.
- Daniel Wilinski
- , Chen Qiu
- & Marvin Wickens
-
Article
| Open Access Large-scale models of signal propagation in human cells derived from discovery phosphoproteomic data
Large-scale models of signal propagation in human cells derived from discovery phosphoproteomic data
Phosphoproteomics can offer significant insight into cell signalling and how signalling is modified in response to perturbations. Here the authors develop a new tool for the analysis of high-content phosphoproteomics in the context of kinase/phosphatase-substrate knowledge, which is used to train logic models.
- Camille D. A. Terfve
- , Edmund H. Wilkes
- & Julio Saez-Rodriguez
-
Article
| Open Access Defining the relationship between infection prevalence and clinical incidence of Plasmodium falciparum malaria
Defining the relationship between infection prevalence and clinical incidence of Plasmodium falciparum malaria
Mathematical models are used to predict malaria burden to inform disease control efforts. Here, Cameron et al. use Bayesian statistics to calibrate previous models against a data set of age-structured prevalence and incidence, generating stratified forecasts of the prevalence–incidence relationship.
- Ewan Cameron
- , Katherine E. Battle
- & Peter W. Gething
-
Article
| Open Access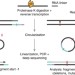 Improved binding site assignment by high-resolution mapping of RNA–protein interactions using iCLIP
Improved binding site assignment by high-resolution mapping of RNA–protein interactions using iCLIP
Individual-nucleotide resolution crosslinking and immunoprecipitation (iCLIP) can map RNA binding sites of RNA-binding proteins (RBPs). Here, the authors report an analysis tool that improves the binding site assignment for some RBPs that have length-dependent broader distribution for their iCLIP fragments.
- Christian Hauer
- , Tomaz Curk
- & Andreas E. Kulozik
-
Article
| Open Access mRIN for direct assessment of genome-wide and gene-specific mRNA integrity from large-scale RNA-sequencing data
mRIN for direct assessment of genome-wide and gene-specific mRNA integrity from large-scale RNA-sequencing data
With the rapid increase in the volume of publically available RNA-seq data, quality control is an increasingly important consideration. Here Feng et al. develop mRIN, a method to directly assess mRNA integrity, and show that RNA degradation in post-mortem samples has a strong impact on global expression profiles.
- Huijuan Feng
- , Xuegong Zhang
- & Chaolin Zhang
-
Article
| Open Access Quantitative interactome analysis reveals a chemoresistant edgotype
Quantitative interactome analysis reveals a chemoresistant edgotype
Changes in protein–protein interactions result in changes to cellular phenotype. Here the authors use crosslinking mass spectrometry to derive a quantitative protein interaction network in drug-sensitive and -resistant HeLa cells, and uncover a chemoresistant ‘edgotype’.
- Juan D. Chavez
- , Devin K. Schweppe
- & James E. Bruce
-
Article
| Open Access RC3H1 post-transcriptionally regulates A20 mRNA and modulates the activity of the IKK/NF-κB pathway
RC3H1 post-transcriptionally regulates A20 mRNA and modulates the activity of the IKK/NF-κB pathway
The RNA-binding protein RC3H1/ROQUIN1 promotes the degradation of mRNA by binding to a consensus CDE present in the 3′UTR. Here the authors expand the set of consensus sequences through which RCH31 binds and regulates mRNA encoding members of the DNA damage response and IKK/NF-κB pathway.
- Yasuhiro Murakawa
- , Michael Hinz
- & Markus Landthaler
-
Article
| Open Access Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA
Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA
Long non-coding RNAs (lncRNAs) have emerged as important regulators of gene expression through several distinct mechanisms. Here the authors further delineate the role of p53-induced lncRNAs within the p53-responsive pathways through the activation of enhancers.
- Nicolas Léveillé
- , Carlos A. Melo
- & Reuven Agami
-
Article |
 Structural and evolutionary versatility in protein complexes with uneven stoichiometry
Structural and evolutionary versatility in protein complexes with uneven stoichiometry
Although many heteromeric protein complexes exhibit 1:1 ratios between their components, a significant number feature uneven stoichiometry. Marsh et al. perform a global analysis of uneven stoichiometry, identifying structural mechanisms by which it is achieved, and explaining its differential conservation.
- Joseph A. Marsh
- , Holly A. Rees
- & Sarah A. Teichmann
-
Article
| Open Access Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity
Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity
Efficacy of anticancer treatments vary across patients, imposing a need for personalized approaches. Here the authors show that responsiveness to chemotherapy can be predicted using tumour explant cultures in a patient-matched microenvironment, coupled with a machine-learning algorithm.
- Biswanath Majumder
- , Ulaganathan Baraneedharan
- & Pradip K. Majumder
-
Article
| Open Access An analytical framework for optimizing variant discovery from personal genomes
An analytical framework for optimizing variant discovery from personal genomes
The standardization of clinical sequencing data generation and analysis is of critical importance. Here, the authors develop the Genome Comparison and Analytic Testing platform to facilitate the development of performance metrics and comparisons of analysis tools for clinical sequencing studies.
- Gareth Highnam
- , Jason J. Wang
- & David Mittelman
-
Article |
 Sequencing of first-strand cDNA library reveals full-length transcriptomes
Sequencing of first-strand cDNA library reveals full-length transcriptomes
Strand-specific RNA-seq (ssRNA-seq) data often lack information on 5′ and 3′ ends of transcripts. Here the authors present a novel method for ssRNA-seq that enables the simultaneous profiling of gene expression, TSSs and polyadenylation sites at near-base resolution with a single library.
- Saurabh Agarwal
- , Todd S. Macfarlan
- & Shigeki Iwase
-
Article
| Open Access Identification of genetic variants associated with alternative splicing using sQTLseekeR
Identification of genetic variants associated with alternative splicing using sQTLseekeR
RNA sequencing has enabled the global analysis of both gene expression levels and splicing events. Here, the authors develop a multivariate approach that is able to identify SNPs that influence splicing, and investigate the overlap of these with functional domains across the genome, including previously identified GWAS signals.
- Jean Monlong
- , Miquel Calvo
- & Roderic Guigó
-
Article
| Open Access Automated monitoring and quantitative analysis of feeding behaviour in Drosophila
Automated monitoring and quantitative analysis of feeding behaviour in Drosophila
Feeding is an important behaviour, but its quantification remains challenging, particularly in small animal models like Drosophila melanogaster. Here the authors describe a method which uses capacitive sensing for automated high-resolution measuring of feeding behaviour in individual flies.
- Pavel M. Itskov
- , José-Maria Moreira
- & Carlos Ribeiro
-
Article |
 Using synthetic templates to design an unbiased multiplex PCR assay
Using synthetic templates to design an unbiased multiplex PCR assay
Immunosequencing enables cost-effective sequencing of repertoires of immune cells, but it often suffers from amplification biases when attempting cell quantification. Here, the authors present a powerful multiplex PCR assay that allows for quantitative and unbiased analysis of frequency of different T cell receptors.
- Christopher S. Carlson
- , Ryan O. Emerson
- & Harlan Robins
-
Article
| Open Access Inferring tumour purity and stromal and immune cell admixture from expression data
Inferring tumour purity and stromal and immune cell admixture from expression data
Tumour biopsies contain contaminating normal cells and these can influence the analysis of tumour samples. In this study, Yoshihara et al.develop an algorithm based on gene expression profiles from The Cancer Genome Atlas to estimate the number of contaminating normal cells in tumour samples.
- Kosuke Yoshihara
- , Maria Shahmoradgoli
- & Roel G.W. Verhaak
-
Article
| Open Access Active learning framework with iterative clustering for bioimage classification
Active learning framework with iterative clustering for bioimage classification
Semi-automated imaging systems help with the task of classifying large numbers of biological images. This study presents a novel framework—CARTA—with an active learning algorithm combined with a genetic algorithm, whose applications include the classification of magnetic resonance imaging of cancer cells.
- Natsumaro Kutsuna
- , Takumi Higaki
- & Seiichiro Hasezawa
-
Article |
 Reliable detection of subclonal single-nucleotide variants in tumour cell populations
Reliable detection of subclonal single-nucleotide variants in tumour cell populations
The detection of subclonal variants in heterogeneous cancer specimens is a challenge due to errors that occur during sequencing. In this study, a statistical algorithm and a sequencing strategy are reported that circumvent this issue and can accurately detect variants at a frequency as low as 1/10,000.
- Moritz Gerstung
- , Christian Beisel
- & Niko Beerenwinkel
-
Article
| Open Access Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa
Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa
Understanding the genetics and physiology of domesticated species is important for crop improvement. By studying natural variation and the phenotypic traits of 413 diverse accessions of rice, Zhao et al. identify many common genetic variants that influence quantitative traits such as seed size and flowering time.
- Keyan Zhao
- , Chih-Wei Tung
- & Susan R. McCouch
-
Article
| Open Access A segmental genomic duplication generates a functional intron
A segmental genomic duplication generates a functional intron
The appearance of a new intron that splits an exon without disrupting the corresponding peptide sequence is a rare event in vertebrate genomes. Hellstenet al.demonstrate that, under certain circumstances, a functional intron can be produced in a single step by segmental genomic duplication.
- Uffe Hellsten
- , Julie L. Aspden
- & Daniel S. Rokhsar
-
Article
| Open Access Deep resequencing reveals excess rare recent variants consistent with explosive population growth
Deep resequencing reveals excess rare recent variants consistent with explosive population growth
To fully catalogue rare genetic variation in humans, many samples need to be examined. In this study, Coventryet al. resequenced two genes, KCNJ11 and HHEX, in 13,715 humans, and concluded that most of the sequence variation arose recently and that variation is greater than expected.
- Alex Coventry
- , Lara M. Bull-Otterson
- & Charles F. Sing
-
Article
| Open Access Second-generation environmental sequencing unmasks marine metazoan biodiversity
Second-generation environmental sequencing unmasks marine metazoan biodiversity
Recent developments in sequencing technologies have provided the opportunity to investigate the biodiversity of ecosystems. Such a metagenomic approach, combined with taxon clustering, is used here to demonstrate that the species richness of a marine community in Scotland is much greater than anticipated.
- Vera G. Fonseca
- , Gary R. Carvalho
- & Simon Creer
-
Article
| Open Access Dynamic evolution of precise regulatory encodings creates the clustered site signature of enhancers
Dynamic evolution of precise regulatory encodings creates the clustered site signature of enhancers
InDrosophila development, DNA enhancers drive gene expression in response to morphogen gradients. Here, Crocker et al. study the evolution of sequences that bind a Dorsal morphogen complex and demonstrate how evolutionary changes in threshold levels have resulted in complex site clustering of DNA elements.
- Justin Crocker
- , Nathan Potter
- & Albert Erives
-
Article
| Open Access Identification of high-quality cancer prognostic markers and metastasis network modules
Identification of high-quality cancer prognostic markers and metastasis network modules
There has been great interest in attempting to identify gene expression signatures that predict cancer survival. In this study a new algorithm is developed to analyse gene expression datasets that accurately classify both ER+ and ER− breast cancers into low- and high-risk groups.
- Jie Li
- , Anne E.G. Lenferink
- & Edwin Wang
 A combined cryo-EM and molecular dynamics approach reveals the mechanism of ErmBL-mediated translation arrest
A combined cryo-EM and molecular dynamics approach reveals the mechanism of ErmBL-mediated translation arrest