
Abstract
Cassava (Manihot esculenta Crantz) is an important staple food crop in Africa and South America; however, ubiquitous deleterious mutations may severely decrease its fitness. To evaluate these deleterious mutations, we constructed a cassava haplotype map through deep sequencing 241 diverse accessions and identified >28 million segregating variants. We found that (i) although domestication has modified starch and ketone metabolism pathways to allow for human consumption, the concomitant bottleneck and clonal propagation have resulted in a large proportion of fixed deleterious amino acid changes, increased the number of deleterious alleles by 26%, and shifted the mutational burden toward common variants; (ii) deleterious mutations have been ineffectively purged, owing to limited recombination in the cassava genome; (iii) recent breeding efforts have maintained yield by masking the most damaging recessive mutations in the heterozygous state but have been unable to purge the mutation burden; such purging should be a key target in future cassava breeding.
Similar content being viewed by others
Main
For millions of people in the tropics, cassava is the third most consumed carbohydrate source, after rice and maize1. Even though cassava was domesticated in Latin America2,3, it has spread widely and has become a major staple crop in Africa. Although its wild progenitor, M. esculenta sp. falbellifolia, reproduces by seed4, cultivated cassava is notably almost exclusively clonally propagated via stem cutting5. The limited number of recombination events in such vegetatively propagated crops may result in an accumulation of deleterious alleles throughout the genome6. Thus, mutation burden in cassava is expected to be more severe than that in sexually propagated species. Deleterious mutations are considered to be at the heart of inbreeding depression7. Even in elite cassava accessions, inbreeding depression is extremely severe, and a single generation of inbreeding may result in a >60% decrease in fresh root yield8,9. In this study, we sought to identify deleterious mutations in cassava populations, with the goal of accelerating cassava breeding by allowing breeders to purge deleterious mutations more efficiently.
We conducted a comprehensive characterization of genetic variation by whole-genome sequencing (WGS) of 241 cassava accessions (Fig. 1, Supplementary Fig. 1 and Supplementary Table 1). On average, more than 30× coverage was generated for each accession. To ensure high-quality variant discovery, variants from low-copy-number regions of the cassava genome10,11 were identified to develop the cassava haplotype map II (HapMapII) (Supplementary Fig. 2), containing 25.9 million SNPs and 1.9 million insertions/deletions (indels) (Supplementary Table 2), of which nearly 50% were rare (minor-allele frequency <0.05) (Supplementary Fig. 3). The error rate of variant calling, i.e., the proportion of segregating sites in the reference accession, was 0.01%. The correlation between read depth and the proportion of SNP heterozygosity was extremely low (r2 = 6 × 10−5). Haplotypes were phased, and missing genotypes were imputed with high accuracy with BEAGLE v4.1 (ref. 12) (accuracy r2 = 0.966) (Supplementary Fig. 4). Linkage disequilibrium was as low as that in maize13 and decayed to an average r2 of 0.1 in 3,000 bp (Supplementary Fig. 5).

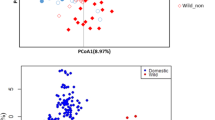
A total of 241 accessions were collected in the study, including 203 elite breeding accessions (M. esculenta Crantz), 16 close relatives (M. esculenta sp. flabelifolia and M. esculenta sp. peruviana) of modern cultivars2,3, 11 hybrid/tree cassava accessions, and 11 more divergent wild relatives (Manihot glaziovii and others). (a) PCA of all cassava accessions (progenitors, cultivated, and wild cassava accessions, n = 241). A total of 43.8% genetic variance was captured in the first two principal components. (b) PCA of cultivated cassava clones (n = 203). A total of 9.1% genetic variance was captured in the first two principal components. LAC, Latin American cassava; ACC, Asian cultivated cassava; EAC, East African cassava; WAC, West African cassava; CWL, crosses between WAC and LAC.
Cultivated cassava had lower nucleotide diversity (pairwise nucleotide diversity (π) = 0.0036) than did its progenitors (M. esculenta sp. flabelifolia, π = 0.0051). In addition, a close relationship between the two species was observed in a phylogenetic analysis (Supplementary Fig. 6). Both lines of evidence supported the hypothesis that cultivated cassava was domesticated from M. esculenta sp. flabelifolia2,3,10. To evaluate population differentiation of cassava, a principal component analysis (PCA) was performed and showed substantial differentiation among all cassava species and hybrids (Fig. 1a): cultivated cassava showed moderate genetic differentiation from its progenitors (fixation index (Fst) = 0.16) and high genetic differentiation from tree cassava (Fst = 0.32) and wild relatives (Fst = 0.44) (Supplementary Table 2 and Supplementary Fig. 7). However, PCA showed very little differentiation among cultivated cassava (Fig. 1b), and cultivated cassava within geographic subpopulations presented unexpectedly low values of Fst (0.01–0.05) even though these subpopulations were sampled from different continents (Supplementary Table 2). This result suggested that despite clonal propagation, there has been sufficient crossing to maintain cultivated cassava in one breeding pool.
Sequence conservation is a powerful tool to discover functional variation14,15. We identified candidate deleterious mutations by using genomic evolution and amino acid conservation modeling. The cassava genome was aligned to seven species in the Malpighiales clade to identify evolutionarily constrained regions in the cassava genome. On the basis of the genomic evolutionary rate profiling (GERP)16 score, nearly 104 Mb of the genome (20%) of cassava was constrained (GERP score >0) (Supplementary Fig. 8). The evolutionarily constrained genome of cassava (104 Mb) was comparable to that of maize (111 Mb)17 in size, but was smaller than that of humans (214 Mb)16 and larger than that of Drosophila (88 Mb)18. GERP profiling also identified a remarkably asymmetric distribution of constrained sequence at the chromosome scale (Supplementary Fig. 9). In addition to the constraint estimation at the DNA level, consequences of mutation on amino acids in proteins were assessed by using the Sorting Intolerant From Tolerant (SIFT) program19. Nearly 3.0% of coding SNPs in cultivated cassava were nonsynonymous mutations (Supplementary Table 2), of which 19.3% (57,952) were putatively deleterious (SIFT <0.05). Because the strength of functional prediction methods varies14, we combined the criteria of SIFT (<0.05) and GERP (>2) to obtain a more conservative set of 22,495 deleterious mutations (Supplementary Fig. 10).
To estimate the individual mutation burden, we used rubber (Hevea brasiliensis), which diverged from the cassava lineage 27 million years (Myr) ago10, as an outgroup to identify derived deleterious alleles in cassava. The derived allele frequency (DAF) spectrum showed that cassava (4.6%, Fig. 2) appeared to have more fixed deleterious mutations than maize (3.2%, DAF >0.8)20 when compared at the same threshold (SIFT <0.05). Across cultivated cassava, there were 150 fixed deleterious mutations. These deleterious mutations cannot be purged through standard breeding, which relies on recombination of segregating alleles, but they are potential targets for genome editing21. Together with the other 22,345 segregating deleterious mutations, the mutation burden in cassava was substantial. Given the several millennia of breeding in the species, why are these deleterious mutations still present in cultivated cassava, and how have breeders been managing them? We evaluated recombination, selection, and drift as the main processes controlling the distribution of deleterious mutations in cassava.

The DAF distribution of alleles is shown. The rubber genome was used as the outgroup to define the derived alleles.
Recombination is an essential process to purge deleterious mutations from the genome22. In vegetatively propagated species such as cassava, recombination is expected be less efficient in purging deleterious mutations. This hypothesis was supported by a weak correlation between the recombination rate and the distribution of deleterious mutations (Pearson's r = −0.066, P = 0.13, Fig. 3a). Deleterious mutations were nearly uniformly spread across the cassava genome (Fig. 3b and Supplementary Fig. 11) rather than being concentrated in low-recombination regions, as seen in humans23, fruit flies24, and maize17. Thus, recombination, which is presumably rare in a clonally propagated crop, does not effectively purge the mutation burden in cassava.

(a) Correlation coefficient between the recombination rate and the number of deleterious mutations in the genome. A total of 529 data points are plotted. The Pearson correlation coefficient (r) was calculated. The significance (P) was determined by two-tailed Student's t test. (b) Distribution of deleterious mutations as a function of recombination rate on chromosome 1.
Domestication is important in the evolution and improvement of crop species. The major domestication trait of cassava is the large carbohydrate-rich storage root. Cultivated cassava has a starch content 5 to 6 times higher than that of its progenitor4. Another domestication trait is the decreased cyanide content in roots4. Every tissue of cassava contains cyanogenic glucosides25. Ketones, cyanohydrin, and hydrogen cyanide are the key toxic compounds formed during degradation of cyanogenic glucosides25,26. These toxic compounds must be eliminated before human consumption. To identify the genomic regions under selection during domestication, the cross-population composite likelihood ratio (XP-CLR)27 was used to scan the genome in Latin American cassava (LAC) accessions and the progenitor (M. esculenta sp. flabelifolia). We identified 203 selective sweeps containing 427 genes in LAC (Supplementary Fig. 12a). Genes in these sweep regions showed enrichment in starch and sucrose synthesis (3.8-fold enrichment; false discovery rate (FDR) = 7.2 × 10−3) and cellular ketone metabolism (3.4-fold enrichment; FDR = 5.3 × 10−3) (Supplementary Fig. 12b). The results suggested that selection during domestication increased the production of carbohydrates and decreased the cyanogenic glucoside content in cassava. Likewise, selection signatures of a recent bottleneck event in African cassava (AC) accessions were also evaluated. A total of 244 selective sweeps were identified, containing 416 genes. These genes were enriched in serine family amino acid metabolism (4.2-fold enrichment, FDR = 2.1 × 10−6) and cellular response to stress (1.3-fold enrichment, FDR = 4.9 × 10−6, Supplementary Fig. 12c,d). Because L-serine is involved in the plant response to biotic and abiotic stresses28,29, together with the functional enrichment in cellular response to stress, this result may reflect that disease-resistance accessions were selected for in a recent breeding program in Africa9.
How was the mutation burden shaped in the selective sweeps? We found that LAC, compared with progenitors, showed 25% fewer (P = 0.009, Fig. 4a) deleterious alleles in sweep regions. Similarly, AC, compared with LAC, exhibited a 35% decrease (P = 2.1 × 10−7, Fig. 4b) in sweep regions. In addition to the comparison among populations, significant within-population decreases in deleterious alleles were observed by comparing sweep regions with the rest of the genome. For example, selective sweeps exhibited a 44% decrease (P = 9.7 × 10−12, Fig. 4c) in deleterious alleles in LAC and a 41% decrease (P = 8.7 × 10−130, Fig. 4d) in AC. This result suggests that haplotypes containing fewer deleterious alleles have been favored during selection.

Box-and-whisker plots were used to compare the mutation burden. Each box represents the mean and interquartile range (IQR). The top whisker denotes the maximum value or the third quartile plus 1.5× the IQR (Q3 + 1.5× IQR), whichever is smaller. The bottom whisker denotes either the minimum value or the first quartile minus 1.5× the IQR (Q1 – 1.5× IQR), whichever is larger. The dots are either more than the third quartile plus 1.5× the IQR or less than the first quartile minus 1.5× the IQR. (a) Mutation burden between progenitors (n = 16) and Latin American cassava accessions (n = 21) in domestication sweep regions. (b) Mutation burden between African (n = 174) and Latin American (n = 21) cassava accessions in sweep regions identified in recent improvement in Africa. (c) Mutation burden in Latin American cassava accessions (n = 21) between domestication selective sweeps and control regions (rest of the genome). CDS, coding DNA sequence. (d) Mutation burden in African cassava accessions between sweep regions identified in recent improvement and control regions (rest of the genome) in Africa.
However, drift after domestication may have played a more important role in affecting mutation burden in cassava. Although LAC and AC, compared with their progenitors, had a similar number of deleterious alleles (P = 0.42, Fig. 5a), they exhibited a prominent increase in total burden by 26% (P = 9.1 × 10−9, Fig. 5a) and a shifted burden toward common deleterious variants (Supplementary Fig. 13). The increase in deleterious alleles during domestication has also been found in dogs30. The results suggest that the severe bottleneck in domestication and the shift from sexual reproduction to clonal propagation have resulted in a rapid accumulation of deleterious alleles in cultivated cassava.

Box-and-whisker plots were used to compare the mutation burden in a–c. The top whisker denotes the maximum value or the third quartile plus 1.5× the IQR (Q3 + 1.5× IQR), whichever is smaller. The bottom whisker denotes either the minimum value or the first quartile minus 1.5× the IQR (Q1 – 1.5× IQR), whichever is larger. The dots are either more than the third quartile plus 1.5× the IQR or less than the first quartile minus 1.5× the IQR. (a) Total mutation burden in progenitors (n = 16), Latin American cassava (n = 21) and African cassava (n = 174) accessions. A bottleneck during domestication increased the mutation burden by 26% (P = 9.1 × 10−9). Demography in Africa had no significant influence on the mutation burden in African cassava accessions (P = 0.42). (b) Homozygous-mutation burden in cassava populations. Domestication decreased the homozygous-mutation burden in cultivated cassava by 23% (P = 7 × 10−3). (c) Heterozygous-mutation burden in cassava populations. Domestication increased the heterozygous-mutation burden in cultivated cassava by 96% (P = 8.1 × 10−7). (d) Comparison between the observed homozygous-mutation burden (n = 10,000) and the expected homozygous-mutation burden (n = 10,000) under the assumption of HWE in cultivated cassava. (e) Comparison between the observed heterozygous-mutation burden (n = 10,000) and the expected heterozygous-mutation burden (n = 10,000) under the assumption of HWE in cultivated cassava.
How have breeders been able to maintain yield, given the substantial increase in mutation burden in cultivated cassava? The answer became apparent when the homozygous and heterozygous deleterious alleles were compared. In cultivated accessions, compared with progenitors, the homozygous-mutation burden substantially decreased, by 23% (P = 7 × 10−3, Fig. 5b), regardless of the elevated frequency of deleterious alleles (Supplementary Fig. 13), whereas the heterozygous-mutation burden markedly increased, by 96% (P = 8.1 × 10−7, Fig. 5c), despite the decreased genetic diversity in cultivated cassava (π = 0.0036) compared with progenitors (π = 0.0051). In addition, we also compared the observed and mutation burdens under the assumption of Hardy–Weinberg Equilibrium (HWE) in cultivated cassava. The relative depletion of the homozygous-mutation burden and the excess heterozygous-mutation burden would not have been present unless they were selected for and maintained. The results showed a decreased homozygous-mutation burden (LAC, 5.6% decrease, P = 0; AC, 10.3% decrease, P = 0, Fig. 5d) and an increased observed heterozygous-mutation burden (LAC, 3.5% increase, P = 1.5 × 10−312; AC, 6.9% increase, P = 0, Fig. 5e), thus indicating a significant deviation from the HWE expectation. These results suggested that breeders have been trying to manage the recessive deleterious mutations in the heterozygous state to mask the harmful effects.
Mutations with a large homozygous effect are more likely to be recessive31. We found that nearly 64.5% of deleterious mutations occurred only in the heterozygous state. Although the low allele frequency prevents effective tests for excess heterozygosity of these deleterious mutations, these mutations are more likely to be strongly deleterious, thus resulting in the significant yield loss in the first generation of selfed cassava plants8,9. These mutations were in genes (n = 7,774) exhibiting functional enrichment in primarily macromolecule catabolism and biosynthesis. In contrast, the deleterious mutations existing predominantly in the homozygous state (proportion of homozygotes >70%) were present in genes (n = 245) exhibiting functional enrichment in amine and ketone metabolism, as well as chemical and stimulus responses (Supplementary Fig. 14).
Using deep sequencing from a comprehensive and representative collection of 241 cassava accessions, we developed HapMapII, a valuable resource for cassava genetic studies and breeding. In this vegetatively propagated species, deleterious mutations have been accumulating rapidly, owing to limited recombination and a domestication bottleneck. Although breeding efforts have successfully maintained yield by selecting high-fitness haplotypes at several hundred loci and handling most damaging mutations in the heterozygous state, breeders have been unable to purge the mutation burden. Instead, they have shielded deleterious mutations by increasing the heterozygosity while screening thousands of potential hybrids (Supplementary Fig. 15). In the short term, this practice for managing mutation burden may produce gains in yield. In the long term, however, a mutational meltdown may be triggered by new mutations, decreasing genetic diversity in the breeding pool, and clonal propagation. Deleterious mutations should be important targets for future genetic research and breeding of cassava. In genetic research, mutations in fast-evolving regulatory regions must be evaluated by examining conservation from closely related species (divergence <5 Myr ago). In addition, dominance effects of deleterious mutations and the interactions among them must be qualified from populations; for breeding, dedicatedly designed crosses and selfing can be applied to eliminate deleterious mutations efficiently. Purging deleterious mutations from cassava, combined with genomic selection and genomic editing technologies21, should improve this globally important crop.
Methods
Samples and whole-genome sequencing.
To maximize the diversity and representation for cassava, all samples were selected on the basis of breeders' choice and diversity analysis from accessions included in Next Generation Cassava Breeding project (URLs). Whole-genome sequences were generated from 241 cassava accessions including 203 elite breeding accessions, 16 progenitors (M. falbellifolia and M. peruviana)2,3, 11 hybrid/tree cassava accessions and 11 wild relative cassava accessions (M. glaziovii and others) (Supplementary Table 1). Wild M. glaziovii has been extensively used in cassava breeding programs to transfer disease-resistance alleles to cultivated cassava (for example, in the Amani Breeding Program)10. Among 241 cassava accessions, 172 accessions were sequenced at the Genomic Diversity Facility at Cornell University. Standard Illumina TruSeq PCR-free libraries were constructed with an insert size of 500 bp. Sequences of 200 bp in length were generated with the Illumina HiSeq 2500 platform, and sequences of 150 bp in length were generated with NextSeq 500 Desktop sequencers. The Donald Danforth Plant Science Center generated ∼20× coverage sequences for 15 elite cassava accessions. Sequences for the remaining 54 cassava accessions were collected from HapMapI10, generated at the University of California, Berkeley.
Alignment of reads and variant calling of cassava haplotype map (HapMapII).
The cassava genome was found to have large amounts of repeat sequences. The 518.5-Mb cassava genome (v6.1) has ∼51% repetitive elements with several common recent retrotransposons10. To exclude misalignment and to ensure high-quality variant discovery, these repeats were prefiltered by aligning the reads to a bait10 containing repeat sequences and organelle sequences (Supplementary Fig. 2). The Burrows–Wheeler alignment with maximal exact matches (BWA-MEM) algorithm32 was used to align and filter repeat reads. We set the --c parameter option to 100,000 to maximize the power to detect repeat reads. Remaining reads after prefiltering were aligned to the reference genome (v6.1)10 with BWA-MEM32 with default parameters. All alignment files were converted to BAM format33. To perform high-quality variant calling and genotyping, especially for rare variants, we developed an in-house pipeline, FastCall (URLs), to perform stringent variant discovery. The alignment records were generated from alignment BAM files with the mpileup tool in Samtools33. The following procedures were included in FastCall: (i) genomic positions with both insertion and deletion variants were ignored, because these sites were probably in complex regions with many misalignments; (ii) for multiple allelic sites, if the third allele had more than 20% depth in any individual, the site was ignored; (iii) for a specific site, if the minor allele did not have a depth between 40% and 60% in at least one individual when the individual depth was greater than 5, the site was ignored; (iv) a chi-squared test for allele segregation13 in all individuals was performed. Sites with P values greater than 1.0 × 10−3 were ignored; (v) On average, over 30× depth was used for individual genotype calls. The genotype likelihood was calculated on the basis of a multinomial test, as previously described34. To remove potential spurious variants arising from paralogs, an additional filter was applied to keep only variants with a depth between 7,500 and 11,500 (Supplementary Fig. 4b). The missing data composed approximately 4%. The genotypes were imputed and phased into haplotypes with BEAGLE v4.1 (ref. 12) with a default window size of 50,000 SNPs.
Error-rate estimates of HapMapII.
The cassava reference accession AM560-2 is a S3-derived inbred11. Therefore, few genetic polymorphisms were expected in the reference genome. The percentage of polymorphic sites across the reference genome was identified as the false-positive error rate of cassava HapMapII (Supplementary Fig. 4a). To estimate imputation accuracy, a total of 10% of the known genotypes (with a minimum read depth of 10) were masked before imputation with BEAGLE. The correlation (Pearson's r) between the imputed genotype and the masked genotype was calculated to evaluate the imputation error.
Population genetic analysis.
SNP density, pairwise nucleotide diversity (π), Tajima's D and the fixation index (Fst) were calculated with VCFtools35 (Supplementary Table 2). SNP density was calculated in 100-kb nonoverlapping windows, Tajima's D and Fst were calculated in 5-kb nonoverlapping windows. Values of π were calculated with variant and invariant sites. PCA was carried out with a distance matrix generated in in Trait Analysis by Association, Evolution and Linkage (TASSEL)36. Phylogenetic analysis was performed with the Analysis of Phylogenetics and Evolution (APE) package37 in R software (Supplementary Fig. 6).
Recombination-rate analysis.
Genetic-linkage-map positions were obtained from the cassava HapMapI source10 and the International Cassava Genetic Map Consortium (ICGMC)38. Genetic-linkage-map positions (in centimorgans) were projected to HapMapII sites through simple linear interpolation between the markers.
Genomic evolutionary rate profiling (GERP).
Constrained portions of the cassava genome were identified by quantifying rejected substitutions (strength of purifying selection) with the GERP++ program16. Multiple whole-genome sequence alignment was carried out for the seven species in the Malpighiales clade of the plant kingdom, including cassava, rubber (H. brasiliensis)39, jatropha (Jatropha curcas)40, castor bean (Ricinus communis)41, willow (Salix purpurea)42, flax (Linum usitatissimum)43, and poplar (Populus trichocarpa)44. Whole-genome alignment was carried out with the Large-Scale Genome Alignment Tool (LASTZ)45. Phylogenetic tree and neutral branch length (estimated from fourfold degenerate sites) analyses were used to quantify the constraint intensity at every position in the cassava genome. Cassava genome sequences were eliminated during the site-specific observed estimates (rejected substitution (RS) scores) to eliminate the confounding influence of deleterious derived alleles segregating in cassava populations present in the reference sequence.
Identifying deleterious mutations.
Amino acid substitutions and their effects on protein function were predicted with the SIFT algorithm19. Nonsynonymous mutations with SIFT scores <0.05 were defined as putative deleterious mutations. SIFT (<0.05) and GERP (>2) annotations were combined to identify the deleterious mutations in constrained portions of the genome. These deleterious mutations were used to calculate the cassava mutation burden.
The rubber genome was used as an outgroup to identify the deleterious alleles in the cassava genome. At a given position, if a cassava reference allele matched the rubber reference allele, the allele in cassava was categorized as an ancient allele. If a cassava allele was different from the rubber allele, the cassava allele was defined as a derived allele. If the cassava genome was not aligned to the rubber genome, or both reference and alternative alleles did not match the rubber genome, that particular site was ignored. Reference accession (inbred, generation S3) and introgression lines were removed during estimation of mutation burden for each accession.
Identifying selective sweep regions.
The cross-population composite likelihood approach (XP-CLR)27 was used to identify the selective sweeps for two comparisons: Latin America cassava accessions (test populations) versus progenitors (M. esculenta sp. flabelifolia, reference population) for domestication events, and African cassava accessions (test populations) versus Latin American cassava accessions (reference population) to assess recent improvement in Africa. A selection scan was performed across the genome with a 0.5-cM sliding window between the SNPs with a spacing of 2 kb. A genetic map of cassava generated by the International Cassava Genetic Map Consortium38 was used in the XP-CLR analysis. XP-CLR scores were normalized with Z scores and a smoothed spline technique in the R package (GenWin)46. Outlier peaks were selected if they were above the ninety-ninth percentile of normalized values. AgriGO47 and REVIGO48 tools were used for gene ontology (GO) enrichment analysis.
Mutation burden in cassava accessions.
The numbers of derived deleterious alleles present in cassava accessions were counted to identify the mutation burden in cassava accessions in three models (homozygous-mutation burden, heterozygous-mutation burden, and total mutation burden). The homozygous-mutation burden is the number of derived deleterious alleles in the homozygous state. The heterozygous-mutation burden is the number of derived deleterious alleles existing in the heterozygous state. The total mutation burden is the number of derived deleterious alleles existing in an accession (2× homozygous-mutation burden + heterozygous-mutation burden)15,49.
Comparison of observed and expected mutation burden under HWE.
A bootstrap approach (with replacement) was used to resample cultivated cassava accessions from both Latin American (24 samples) and African (174 samples) breeding pools. The process was repeated 10,000 times to generate the distribution of expected homozygous and heterozygous-mutation burden. For each resampling,

where bho is the expected homozygous-mutation burden under HWE, bhe is the expected heterozygous-mutation burden under HWE, n is the total number of deleterious mutations identified (n = 22,495), and di is the allele frequency of the ith deleterious allele in the sampled population. The observed mutation burden was calculated for each accession, as described in the section 'Mutation burden in cassava accessions'. The means of observed homozygous and heterozygous mutation were used for the comparison.
Statistical tests.
The significance of the Pearson correlation coefficient (r) was determined by two-tailed Student's t tests. The difference between groups was tested by unpaired two-tailed Welch's t tests, assuming unequal variance between groups. n represents the sample size.
Data availability.
Whole-genome sequences, and raw and imputed HapMapII SNPs can be accessed through CassavaBase at ftp://ftp.cassavabase.org/HapMapII/.
URLs.
Next Generation Cassava Breeding project, http://www.nextgencassava.org/; FastCall, https://github.com/Fei-Lu/FastCall/.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Raven, P., Fauquet, C., Swaminathan, M.S., Borlaug, N. & Samper, C. Where next for genome sequencing? Science 311, 468–468 (2006).
Olsen, K.M. & Schaal, B.A. Evidence on the origin of cassava: phylogeography of Manihot esculenta. Proc. Natl. Acad. Sci. USA 96, 5586–5591 (1999).
Allem, A.C. The closest wild relatives of cassava (Manihot esculenta Crantz). Euphytica 107, 123–133 (1999).
Wang, W. et al. Cassava genome from a wild ancestor to cultivated varieties. Nat. Commun. 5, 5110 (2014).
McDonald, M.J., Rice, D.P. & Desai, M.M. Sex speeds adaptation by altering the dynamics of molecular evolution. Nature 531, 233–236 (2016).
McKey, D., Elias, M., Pujol, B. & Duputié, A. The evolutionary ecology of clonally propagated domesticated plants. New Phytol. 186, 318–332 (2010).
Charlesworth, D. & Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 10, 783–796 (2009).
Rojas, M.C. et al. Analysis of inbreeding depression in eight s1 cassava families. Crop Sci. 49, 543–548 (2009).
Nuwamanya, E., Herselman, L. & Ferguson, M. Segregation of selected agronomic traits in six S1 cassava families. J. Plant Breed. Crop Sci. 3, 154–160 (2011).
Bredeson, J.V. et al. Sequencing wild and cultivated cassava and related species reveals extensive interspecific hybridization and genetic diversity. Nat. Biotechnol. 34, 562–570 (2016).
Prochnik, S. et al. The cassava genome: current progress, future directions. Trop. Plant Biol. 5, 88–94 (2012).
Browning, B.L. & Browning, S.R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 98, 116–126 (2016).
Chia, J.-M. et al. Maize HapMap2 identifies extant variation from a genome in flux. Nat. Genet. 44, 803–807 (2012).
Tennessen, J.A. et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69 (2012).
Fu, W. et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220 (2013).
Davydov, E.V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP. PLoS Comput. Biol. 6, e1001025 (2010).
Rodgers-Melnick, E. et al. Recombination in diverse maize is stable, predictable, and associated with genetic load. Proc. Natl. Acad. Sci. USA 112, 3823–3828 (2015).
Mackay, T.F.C. et al. The Drosophila melanogaster Genetic Reference Panel. Nature 482, 173–178 (2012).
Kumar, P., Henikoff, S. & Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Mezmouk, S. & Ross-Ibarra, J. The pattern and distribution of deleterious mutations in maize. G3 (Bethesda) 4, 163–171 (2014).
Horvath, P. & Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170 (2010).
Keller, P.J. & Knop, M. Evolution of mutational robustness in the yeast genome: a link to essential genes and meiotic recombination hotspots. PLoS Genet. 5, e1000533 (2009).
Hussin, J.G. et al. Recombination affects accumulation of damaging and disease-associated mutations in human populations. Nat. Genet. 47, 400–404 (2015).
Haddrill, P.R., Halligan, D.L., Tomaras, D. & Charlesworth, B. Reduced efficacy of selection in regions of the Drosophila genome that lack crossing over. Genome Biol. 8, R18 (2007).
Jørgensen, K. et al. Cassava plants with a depleted cyanogenic glucoside content in leaves and tubers: distribution of cyanogenic glucosides, their site of synthesis and transport, and blockage of the biosynthesis by RNA interference technology. Plant Physiol. 139, 363–374 (2005).
Conn, E.E. Cyanogenic compounds. Annu. Rev. Plant Physiol. 31, 433–451 (1980).
Chen, H., Patterson, N. & Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 20, 393–402 (2010).
Ros, R., Muñoz-Bertomeu, J. & Krueger, S. Serine in plants: biosynthesis, metabolism, and functions. Trends Plant Sci. 19, 564–569 (2014).
Benstein, R.M. et al. Arabidopsis phosphoglycerate dehydrogenase1 of the phosphoserine pathway is essential for development and required for ammonium assimilation and tryptophan biosynthesis. Plant Cell 25, 5011–5029 (2013).
Marsden, C.D. et al. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc. Natl. Acad. Sci. USA 113, 152–157 (2016).
Agrawal, A.F. & Whitlock, M.C. Inferences about the distribution of dominance drawn from yeast gene knockout data. Genetics 187, 553–566 (2011).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997/ (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Hohenlohe, P.A. et al. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 6, e1000862 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Bradbury, P.J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Paradis, E., Claude, J. & Strimmer, K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004).
International Cassava Genetic Map Consortium (ICGMC). High-resolution linkage map and chromosome-scale genome assembly for cassava (Manihot esculenta Crantz) from 10 populations. G3 (Bethesda) 5, 133–144 (2015).
Rahman, A.Y.A. et al. Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genomics 14, 75 (2013).
Sato, S. et al. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res. 18, 65–76 (2011).
Chan, A.P. et al. Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 28, 951–956 (2010).
Dai, X. et al. The willow genome and divergent evolution from poplar after the common genome duplication. Cell Res. 24, 1274–1277 (2014).
Wang, Z. et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 72, 461–473 (2012).
Tuskan, G.A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604 (2006).
Harris, R.S. Improved pairwise alignment of genomic DNA. PhD thesis, Pennsylvania State University (2007).
Beissinger, T.M., Rosa, G.J., Kaeppler, S.M., Gianola, D. & de Leon, N. Defining window-boundaries for genomic analyses using smoothing spline techniques. Genet. Sel. Evol. 47, 30 (2015).
Du, Z., Zhou, X., Ling, Y., Zhang, Z. & Su, Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70 (2010).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800 (2011).
Henn, B.M. et al. Distance from sub-Saharan Africa predicts mutational load in diverse human genomes. Proc. Natl. Acad. Sci. USA 113, E440–E449 (2016).
Acknowledgements
This work was supported by the Bill & Melinda Gates Foundation (BMGF 01511000147 (E.S.B.)), with additional support from the NSF Plant Genome Research Project (1238014 (E.S.B.)) and the UDSA-ARS. We thank the Next Generation Cassava Breeding project for helping us select the accessions to include in WGS efforts. We thank S.E. Prochnik (DOE Joint Genome Institute) for timely assistance during the analysis.
Author information
Authors and Affiliations
Contributions
The manuscript was prepared by P.R. and F.L. Data analysis was carried out by P.R., F.L., and J.V. Whole-genome sequences for 54 accessions included in HapMapI10 were provided by J.V.B., W.E., I.Y.R., C.E., and R.K.; R.S.B. provided the germplasm for WGS. All authors provided comments and edited the manuscript. F.L. and E.S.B. designed and coordinated the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–15 and Supplementary Tables 1 and 2 (PDF 13612 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) License. The images or other third party material in this article is included in the article’s Creative Commons license, unless otherwise indicated in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ramu, P., Esuma, W., Kawuki, R. et al. Cassava haplotype map highlights fixation of deleterious mutations during clonal propagation. Nat Genet 49, 959–963 (2017). https://doi.org/10.1038/ng.3845
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3845
This article is cited by
-
Allelic variation in the autotetraploid potato: genes involved in starch and steroidal glycoalkaloid metabolism as a case study
BMC Genomics (2024)
-
Diploid Interspecific Recombinant Inbred Lines for Genetic Mapping in Potato
American Journal of Potato Research (2024)
-
Metabolic GWAS-based dissection of genetic basis underlying nutrient quality variation and domestication of cassava storage root
Genome Biology (2023)
-
Floral scent of the Mediterranean fig tree: significant inter-varietal difference but strong conservation of the signal responsible for pollinator attraction
Scientific Reports (2023)
-
Population genomics unravels the Holocene history of bread wheat and its relatives
Nature Plants (2023)
