
Abstract
Three subfamilies of grasses, the Ehrhartoideae, Panicoideae and Pooideae, provide the bulk of human nutrition and are poised to become major sources of renewable energy. Here we describe the genome sequence of the wild grass Brachypodium distachyon (Brachypodium), which is, to our knowledge, the first member of the Pooideae subfamily to be sequenced. Comparison of the Brachypodium, rice and sorghum genomes shows a precise history of genome evolution across a broad diversity of the grasses, and establishes a template for analysis of the large genomes of economically important pooid grasses such as wheat. The high-quality genome sequence, coupled with ease of cultivation and transformation, small size and rapid life cycle, will help Brachypodium reach its potential as an important model system for developing new energy and food crops.
Similar content being viewed by others


Main
Grasses provide the bulk of human nutrition, and highly productive grasses are promising sources of sustainable energy1. The grass family (Poaceae) comprises over 600 genera and more than 10,000 species that dominate many ecological and agricultural systems2,3. So far, genomic efforts have largely focused on two economically important grass subfamilies, the Ehrhartoideae (rice) and the Panicoideae (maize, sorghum, sugarcane and millets). The rice4 and sorghum5 genome sequences and a detailed physical map of maize6 showed extensive conservation of gene order5,7 and both ancient and relatively recent polyploidization.
Most cool season cereal, forage and turf grasses belong to the Pooideae subfamily, which is also the largest grass subfamily. The genomes of many pooids are characterized by daunting size and complexity. For example, the bread wheat genome is approximately 17,000 megabases (Mb) and contains three independent genomes8. This has prohibited genome-scale comparisons spanning the three most economically important grass subfamilies.
Brachypodium, a member of the Pooideae subfamily, is a wild annual grass endemic to the Mediterranean and Middle East9 that has promise as a model system. This has led to the development of highly efficient transformation10,11, germplasm collections12,13,14, genetic markers14, a genetic linkage map15, bacterial artificial chromosome (BAC) libraries16,17, physical maps18 (M.F., unpublished observations), mutant collections (http://brachypodium.pw.usda.gov, http://www.brachytag.org), microarrays and databases (http://www.brachybase.org, http://www.phytozome.net, http://www.modelcrop.org, http://mips.helmholtz-muenchen.de/plant/index.jsp) that are facilitating the use of Brachypodium by the research community. The genome sequence described here will allow Brachypodium to act as a powerful functional genomics resource for the grasses. It is also an important advance in grass structural genomics, permitting, for the first time, whole-genome comparisons between members of the three most economically important grass subfamilies.
Genome sequence assembly and annotation
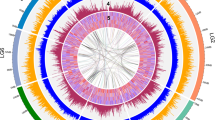
The diploid inbred line Bd21 (ref. 19) was sequenced using whole-genome shotgun sequencing (Supplementary Table 1). The ten largest scaffolds contained 99.6% of all sequenced nucleotides (Supplementary Table 2). Comparison of these ten scaffolds with a genetic map (Supplementary Fig. 1) detected two false joins and created a further seven joins to produce five pseudomolecules that spanned 272 Mb (Supplementary Table 3), within the range measured by flow cytometry20,21. The assembly was confirmed by cytogenetic analysis (Supplementary Fig. 2) and alignment with two physical maps and sequenced BACs (Supplementary Data). More than 98% of expressed sequence tags (ESTs) mapped to the sequence assembly, consistent with a near-complete genome (Supplementary Table 4 and Supplementary Fig. 3). Compared to other grasses, the Brachypodium genome is very compact, with retrotransposons concentrated at the centromeres and syntenic breakpoints (Fig. 1). DNA transposons and derivatives are broadly distributed and primarily associated with gene-rich regions.

The abundance and distribution of the following genome elements are shown: complete LTR retroelements (cLTRs); solo-LTRs (sLTRs); potentially autonomous DNA transposons that are not miniature inverted-repeat transposable elements (MITEs) (DNA-TEs); MITEs; gene exons (CDS); gene introns and satellite tandem arrays (STA). Graphs are from 0 to 100 per cent base-pair (%bp) coverage of the respective window. The heat map tracks have different ranges and different maximum (max) pseudocolour levels: STA (0–55, scaled to max 10) %bp; cLTRs (0–36, scaled to max 20) %bp; sLTRs (0–4) %bp; DNA-TEs (0–20) %bp; MITEs (0–22) %bp; CDS (exons) (0–22.3) %bp. The triangles identify syntenic breakpoints.
We analysed small RNA populations from inflorescence tissues with deep Illumina sequencing, and mapped them onto the genome sequence (Fig. 2a, Supplementary Fig. 4 and Supplementary Table 5). Small RNA reads were most dense in regions of high repeat density, similar to the distribution reported in Arabidopsis22. We identified 413 and 198 21- and 24-nucleotide phased short interfering RNA (siRNA) loci, respectively. Using the same algorithm, the only phased loci identified in Arabidopsis were five of the eight trans-acting siRNA loci, and none was 24-nucelotide phased. The biological functions of these clusters of Brachypodium phased siRNAs, which account for a significant number of small RNAs that map outside repeat regions, are not known at present.

a, Genome-wide distribution of small RNA loci and transcripts in the Brachypodium genome. Brachypodium chromosomes (1–5) are shown at the top. Total small RNA reads (black lines) and total small RNA loci (red lines) are shown on the top panel. Histograms plot 21-nucleotide (nt) (blue) or 24-nucleotide (red) small RNA reads normalized for repeated matches to the genome. The phased loci histograms plot the position and phase-score of 21-nucleotide (blue) and 24-nucleotide (red) phased small RNA loci. Repeat-normalized RNA-seq read histograms plot the abundance of reads matching RNA transcripts (green), normalized for ambiguous matches to the genome. b, Transcript coverage over gene features. Perfect match 32-base oligonucleotide Illumina reads were mapped to the Brachypodium v1.0 annotation features using HashMatch (http://mocklerlab-tools.cgrb.oregonstate.edu/). Plots of Illumina coverage were calculated as the percentage of bases along the length of the sequence feature supported by Illumina reads for the indicated gene model features. The bottom and top of the box represent the 25th and 75th quartiles, respectively. The white line is the median and the red diamonds denote the mean. SJS, splice junction site. c, Venn diagram showing the distribution of shared gene families between representatives of Ehrhartoideae (rice RAP2), Panicoideae (sorghum v1.4) and Pooideae (Brachypodium v1.0, and Triticum aestivum and Hordeum vulgare TCs (transcript consensus)/EST sequences). Paralogous gene families were collapsed in these data sets.
A total of 25,532 protein-coding gene loci was predicted in the v1.0 annotation (Supplementary Information and Supplementary Table 6). This is in the same range as rice (RAP2, 28,236)23 and sorghum (v1.4, 27,640)5, suggesting similar gene numbers across a broad diversity of grasses. Gene models were evaluated using ∼10.2 gigabases (Gb) of Illumina RNA-seq data (Supplementary Fig. 5)24. Overall, 92.7% of predicted coding sequences (CDS) were supported by Illumina data (Fig. 2b), demonstrating the high accuracy of the Brachypodium gene predictions. These gene models are available from several databases (such as http://www.brachybase.org, http://www.phytozome.net, http://www.modelcrop.org and http://mips.org).
Between 77 and 84% of gene families (defined according to Supplementary Fig. 6) are shared among the three grass subfamilies represented by Brachypodium, rice and sorghum, reflecting a relatively recent common origin (Fig. 2c). Grass-specific genes include transmembrane receptor protein kinases, glycosyltransferases, peroxidases and P450 proteins (Supplementary Table 7B). The Pooideae-specific gene set contains only 265 gene families (Supplementary Table 7C) comprising 811 genes (1,400 including singletons). Genes enriched in grasses were significantly more likely to be contained in tandem arrays than random genes, demonstrating a prominent role for tandem gene expansion in the evolution of grass-specific genes (Supplementary Fig. 7 and Supplementary Table 8).
To validate and improve the v1.0 gene models, we manually annotated 2,755 gene models from 97 diverse gene families (Supplementary Tables 9–11) relevant to bioenergy and food crop improvement. We annotated 866 genes involved in cell wall biosynthesis/modification and 948 transcription factors from 16 families25. Only 13% of the gene models required modification and very few pseudogenes were identified, demonstrating the accuracy of the v1.0 annotation. Phylogenetic trees for 62 gene families were constructed using genes from rice, Arabidopsis, sorghum and poplar. In nearly all cases, Brachypodium genes had a similar distribution to rice and sorghum, demonstrating that Brachypodium is suitably generic for grass functional genomics research (Supplementary Figs 8 and 9). Analysis of the predicted secretome identified substantial differences in the distribution of cell wall metabolism genes between dicots and grasses (Supplementary Tables 12, 13 and Supplementary Fig. 10), consistent with their different cell walls26. Signal peptide probability curves also suggested that start codons were accurately predicted (Supplementary Fig. 11).
Maintaining a small grass genome size
Exhaustive analysis of transposable elements (Supplementary Information and Supplementary Table 14) showed retrotransposon sequences comprise 21.4% of the genome, compared to 26% in rice, 54% in sorghum, and more than 80% in wheat27. Thirteen retroelement sets were younger than 20,000 years, showing a recent activation compared to rice28 (Supplementary Fig. 12), and a further 53 retroelement sets were less than 0.1 million years (Myr) old. A minimum of 17.4 Mb has been lost by long terminal repeat (LTR)–LTR recombination, demonstrating that retroelement expansion is countered by removal through recombination. In contrast, retroelements persist for very long periods of time in the closely related Triticeae28.
DNA transposons comprise 4.77% of the Brachypodium genome, within the range found in other grass genomes5,29. Transcriptome data and structural analysis suggest that many non-autonomous Mariner DTT and Harbinger elements recruit transposases from other families. Two CACTA DTC families (M and N) carried five non-element genes, and the Harbinger U family has amplified a NBS-LRR gene family (Supplementary Figs 13 and 14), adding it to the group of transposable elements implicated in gene mobility30,31. Centromeric regions were characterized by low gene density, characteristic repeats and retroelement clusters (Supplementary Fig. 15). Other repeat classes are described in Supplementary Table 15. Conserved non-coding sequences are described in Supplementary Fig. 16.
Whole-genome comparison of three diverse grass genomes
The evolutionary relationships between Brachypodium, sorghum, rice and wheat were assessed by measuring the mean synonymous substitution rates (Ks) of orthologous gene pairs (Supplementary Information, Supplementary Fig. 17 and Supplementary Table 16), from which divergence times of Brachypodium from wheat 32–39 Myr ago, rice 40–53 Myr ago, and sorghum 45–60 Myr ago (Fig. 3a) were estimated. The Ks of orthologous gene pairs in the intragenomic Brachypodium duplications (Fig. 3b) suggests duplication 56–72 Myr ago, before the diversification of the grasses. This is consistent with previous evolutionary histories inferred from a small number of genes3,32,33,34.

a, The distribution maxima of mean synonymous substitution rates (Ks) of Brachypodium, rice, sorghum and wheat orthologous gene pairs (Supplementary Table 16) were used to define the divergence times of these species and the age of interchromosomal duplications in Brachypodium. WGD, whole-genome duplication. The numbers refer to the predicted divergence times measured as Myr ago by the NG or ML methods. b, Diagram showing the six major interchromosomal Brachypodium duplications, defined by 723 paralogous relationships, as coloured bands linking the five chromosomes. c, Identification of chromosome relationships between the Brachypodium, rice and sorghum genomes. Orthologous relationships between the 25,532 protein-coding Brachypodium genes, 7,216 sorghum orthologues (12 syntenic blocks), and 8,533 rice orthologues (12 syntenic blocks) were defined. Sets of collinear orthologous relationships are represented by a coloured band according to each Brachypodium chromosome (blue, chromosome (chr.) 1; yellow, chr. 2; violet, chr. 3; red, chr. 4; green, chr. 5). The white region in each Brachypodium chromosome represents the centromeric region. d, Orthologous gene relationships between Brachypodium and barley and Ae. tauschii were identified using genetically mapped ESTs. 2,516 orthologous relationships defined 12 syntenic blocks. These are shown as coloured bands. e, Orthologous gene relationships between Brachypodium and hexaploid bread wheat defined by 5,003 ESTs mapped to wheat deletion bins. Each set of orthologous relationships is represented by a band that is evenly spread across each deletion interval on the wheat chromosomes.
Paralogous relationships among Brachypodium chromosomes showed six major chromosomal duplications covering 92.1% of the genome (Fig. 3b), representing ancestral whole-genome duplication35. Using the rice and sorghum genome sequences, genetic maps of barley36 and Aegilops tauschii (the D genome donor of hexaploid wheat)37, and bin-mapped wheat ESTs38,39, 21,045 orthologous relationships between Brachypodium, rice, sorghum and Triticeae were identified (Supplementary Information). These identified 59 blocks of collinear genes covering 99.2% of the Brachypodium genome (Fig. 3c–e). The orthologous relationships are consistent with an evolutionary model that shaped five Brachypodium chromosomes from a five-chromosome ancestral genome by a 12-chromosome intermediate involving seven major chromosome fusions39 (Supplementary Fig. 18). These collinear blocks of orthologous genes provide a robust and precise sequence framework for understanding grass genome evolution and aiding the assembly of sequences from other pooid grasses. We identified 14 major syntenic disruptions between Brachypodium and rice/sorghum that can be explained by nested insertions of entire chromosomes into centromeric regions (Fig. 4a, b)2,37,40. Similar nested insertions in sorghum37 and barley (Fig. 4c, d) were also identified. Centromeric repeats and peaks in retroelements at the junctions of chromosome insertions are footprints of these insertion events (Supplementary Fig. 15C and Fig. 1), as is higher gene density at the former distal regions of the inserted chromosomes (Fig. 1). Notably, the reduction in chromosome number in Brachypodium and wheat occurred independently because none of the chromosome fusions are shared by Brachypodium and the Triticeae37 (Supplementary Fig. 18).

a, The five Brachypodium chromosomes are coloured according to homology with rice chromosomes (Os1–Os12). Chromosomes descended from an ancestral chromosome (A4–A11) through whole-genome duplication are shown in shades of the same colour. Gene density is indicated as a red line above the chromosome maps. Major discontinuities in gene density identify syntenic breakpoints, which are marked by a diamond. White diamonds identify fusion points containing remnant centromeric repeats. b, A pattern of nested insertions of whole chromosomes into centromeric regions explains the observed syntenic break points. Bd5 has not undergone chromosome fusion. c, Examples of nested chromosome insertions in sorghum (Sb) chromosomes 1 and 2. d, Examples of nested chromosome insertions in barley (H chromosomes) inferred from genetic maps. Nested insertions were not identified in other chromosomes, possibly owing to the low resolution of genetic maps.
Comparisons of evolutionary rates between Brachypodium, sorghum, rice and Ae. tauschii demonstrated a substantially higher rate of genome change in Ae. tauschii (Supplementary Table 17). This may be due to retroelement activity that increases syntenic disruptions, as proposed for chromosome 5S later41. Among seven relatively large gene families, four were highly syntenic and two (NBS-LRR and F-box) were almost never found in syntenic order when compared to rice and sorghum (Supplementary Table 18), consistent with the rapid diversification of the NBS-LRR and F-box gene families42.
The short arm of chromosome 5 (Bd5S) has a gene density roughly half of the rest of the genome, high LTR retrotransposon density, the youngest intact Gypsy elements and the lowest solo LTR density. Thus, unlike the rest of the Brachypodium genome, Bd5S is gaining retrotransposons by replication and losing fewer by recombination. Syntenic regions of rice (Os4S) and sorghum (Sb6S) demonstrate maintenance of this high repeat content for ∼50–70 Myr (Supplementary Fig. 19)43. Bd5S, Os4S and Sb6S also have the lowest proportion of collinear genes (Fig. 4a and Supplementary Fig. 19). We propose that the chromosome ancestral to Bd5S reached a tipping point in which high retrotransposon density had deleterious effects on genes.
Discussion
As the first genome sequence of a pooid grass, the Brachypodium genome aids genome analysis and gene identification in the large and complex genomes of wheat and barley, two other pooid grasses that are among the world’s most important crops. The very high quality of the Brachypodium genome sequence, in combination with those from two other grass subfamilies, enabled reconstruction of chromosome evolution across a broad diversity of grasses. This analysis contributes to our understanding of grass diversification by explaining how the varying chromosome numbers found in the major grass subfamilies derive from an ancestral set of five chromosomes by nested insertions of whole chromosomes into centromeres. The relatively small genome of Brachypodium contains many active retroelement families, but recombination between these keeps genome expansion in check. The short arm of chromosome 5 deviates from the rest of the genome by exhibiting a trend towards genome expansion through increased retroelement numbers and disruption of gene order more typical of the larger genomes of closely related grasses.
Grass crop improvement for sustainable fuel44 and food45 production requires a substantial increase in research in species such as Miscanthus, switchgrass, wheat and cool season forage grasses. These considerations have led to the rapid adoption of Brachypodium as an experimental system for grass research. The similarities in gene content and gene family structure between Brachypodium, rice and sorghum support the value of Brachypodium as a functional genomics model for all grasses. The Brachypodium genome sequence analysis reported here is therefore an important advance towards securing sustainable supplies of food, feed and fuel from new generations of grass crops.
Methods Summary
Genome sequencing and assembly
Sanger sequencing was used to generate paired-end reads from 3 kb, 8 kb, fosmid (35 kb) and BAC (100 kb) clones to generate 9.4× coverage (Supplementary Table 1). The final assembly of 83 scaffolds covers 271.9 Mb (Supplementary Table 3). Sequence scaffolds were aligned to a genetic map to create pseudomolecules covering each chromosome (Supplementary Figs 1 and 2).
Protein-coding gene annotation
Gene models were derived from weighted consensus prediction from several ab initio gene finders, optimal spliced alignments of ESTs and transcript assemblies, and protein homology. Illumina transcriptome sequence was aligned to predicted genome features to validate exons, splice sites and alternatively spliced transcripts.
Repeats analysis
The MIPS ANGELA pipeline was used to integrate analyses from expert groups. LTR-STRUCT and LTR-HARVEST46 were used for de novo retroelement searches.
Accession codes
Primary accessions
DDBJ/GenBank/EMBL
GenBank/EMBL/DDBJ
Data deposits
The whole-genome shotgun sequence of Brachypodium distachyon has been deposited at DDBJ/EMBL/GenBank under the accession ADDN00000000. (The version described in this manuscript is the first version, accession ADDN01000000). EST sequences have been deposited with dbEST (accessions 67946317–68053959) and GenBank (accessions GT758162–GT865804). The short read archive accession for RNA-seq data is SRA010177.
References
Somerville, C. The billion-ton biofuels vision. Science 312, 1277 (2006)
Kellogg, E. A. Evolutionary history of the grasses. Plant Physiol. 125, 1198–1205 (2001)
Gaut, B. S. Evolutionary dynamics of grass genomes. New Phytol. 154, 15–28 (2002)
International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 436, 793–800 (2005)
Paterson, A. H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009)
Wei, F. et al. Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet. 3, e123 (2007)
Moore, G., Devos, K. M., Wang, Z. & Gale, M. D. Cereal genome evolution. Grasses, line up and form a circle. Curr. Biol. 5, 737–739 (1995)
Salamini, F., Ozkan, H., Brandolini, A., Schafer-Pregl, R. & Martin, W. Genetics and geography of wild cereal domestication in the near east. Nature Rev. Genet. 3, 429–441 (2002)
Draper, J. et al. Brachypodium distachyon. A new model system for functional genomics in grasses. Plant Physiol. 127, 1539–1555 (2001)
Vain, P. et al. Agrobacterium-mediated transformation of the temperate grass Brachypodium distachyon (genotype Bd21) for T-DNA insertional mutagenesis. Plant Biotechnol. J. 6, 236–245 (2008)
Vogel, J. & Hill, T. High-efficiency Agrobacterium-mediated transformation of Brachypodium distachyon inbred line Bd21–3. Plant Cell Rep. 27, 471–478 (2008)
Vogel, J. P., Garvin, D. F., Leong, O. M. & Hayden, D. M. Agrobacterium-mediated transformation and inbred line development in the model grass Brachypodium distachyon . Plant Cell Tissue Organ Cult. 84, 100179–100191 (2006)
Filiz, E. et al. Molecular, morphological and cytological analysis of diverse Brachypodium distachyon inbred lines. Genome 52, 876–890 (2009)
Vogel, J. P. et al. Development of SSR markers and analysis of diversity in Turkish populations of Brachypodium distachyon . BMC Plant Biol. 9, 88 (2009)
Garvin, D. F. et al. An SSR-based genetic linkage map of the model grass Brachypodium distachyon . Genome 53, 1–13 (2009)
Huo, N. et al. Construction and characterization of two BAC libraries from Brachypodium distachyon, a new model for grass genomics. Genome 49, 1099–1108 (2006)
Huo, N. et al. The nuclear genome of Brachypodium distachyon: analysis of BAC end sequences. Funct. Integr. Genomics 8, 135–147 (2008)
Gu, Y. Q. et al. A BAC-based physical map of Brachypodium distachyon and its comparative analysis with rice and wheat. BMC Genomics 10, 496 (2009)
Garvin, D. F. et al. Development of genetic and genomic research resources for Brachypodium distachyon, a new model system for grass crop research. Crop Sci. 48, S-69–S-84 (2008)
Bennett, M. D. & Leitch, I. J. Nuclear DNA amounts in angiosperms: progress, problems and prospects. Ann. Bot. (Lond.) 95, 45–90 (2005)
Vogel, J. P. et al. EST sequencing and phylogenetic analysis of the model grass Brachypodium distachyon . Theor. Appl. Genet. 113, 186–195 (2006)
Rajagopalan, R., Vaucheret, H., Trejo, J. & Bartel, D. P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 20, 3407–3425 (2006)
Tanaka, T. et al. The rice annotation project database (RAP-DB): 2008 update. Nucleic Acids Res. 36, D1028–D1033 (2008)
Fox, S., Filichkin, S. & Mockler, T. Applications of ultra-high-throughput sequencing. Methods Mol. Biol. 553, 79–108 (2009)
Gray, J. et al. A recommendation for naming transcription factor proteins in the grasses. Plant Physiol. 149, 4–6 (2009)
Vogel, J. Unique aspects of the grass cell wall. Curr. Opin. Plant Biol. 11, 301–307 (2008)
Bennetzen, J. L. & Kellogg, E. A. Do plants have a one-way ticket to genomic obesity? Plant Cell 9, 1509–1514 (1997)
Wicker, T. & Keller, B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 17, 1072–1081 (2007)
Wicker, T. et al. Analysis of intraspecies diversity in wheat and barley genomes identifies breakpoints of ancient haplotypes and provides insight into the structure of diploid and hexaploid triticeae gene pools. Plant Physiol. 149, 258–270 (2009)
Jiang, N., Bao, Z., Zhang, X., Eddy, S. R. & Wessler, S. R. Pack-MULE transposable elements mediate gene evolution in plants. Nature 431, 569–573 (2004)
Morgante, M. et al. Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nature Genet. 37, 997–1002 (2005)
Grass Phylogeny Working Group. Phylogeny and subfamilial classification of the grasses (Poaceae). Ann. Mo. Bot. Gard. 88, 373–457 (2001)
Bossolini, E., Wicker, T., Knobel, P. A. & Keller, B. Comparison of orthologous loci from small grass genomes Brachypodium and rice: implications for wheat genomics and grass genome annotation. Plant J. 49, 704–717 (2007)
Charles, M. et al. Sixty million years in evolution of soft grain trait in grasses: emergence of the softness locus in the common ancestor of Pooideae and Ehrhartoideae, after their divergence from Panicoideae . Mol. Biol. Evol. 26, 1651–1661 (2009)
Paterson, A. H., Bowers, J. E. & Chapman, B. A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl Acad. Sci. USA 101, 9903–9908 (2004)
Stein, N. et al. A 1,000-loci transcript map of the barley genome: new anchoring points for integrative grass genomics. Theor. Appl. Genet. 114, 823–839 (2007)
Luo, M. C. et al. Genome comparisons reveal a dominant mechanism of chromosome number reduction in grasses and accelerated genome evolution in Triticeae. Proc. Natl Acad. Sci. USA 106, 15780–15785 (2009)
Qi, L. L. et al. A chromosome bin map of 16,000 expressed sequence tag loci and distribution of genes among the three genomes of polyploid wheat. Genetics 168, 701–712 (2004)
Salse, J. et al. Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell 20, 11–24 (2008)
Srinivasachary, M. M., Gale, M. D. & Devos, K. M. Comparative analyses reveal high levels of conserved colinearity between the finger millet and rice genomes. Theor. Appl. Genet. 115, 489–499 (2007)
Vicient, C. M., Kalendar, R. & Schulman, A. H. Variability, recombination, and mosaic evolution of the barley BARE-1 retrotransposon. J. Mol. Evol. 61, 275–291 (2005)
Meyers, B. C., Kozik, A., Griego, A., Kuang, H. & Michelmore, R. W. Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis . Plant Cell 15, 809–834 (2003)
Ma, J. & Bennetzen, J. L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl Acad. Sci. USA 101, 12404–12410 (2004)
U.S. Department of Energy Office of Science. Breaking the Biological Barriers to Cellulosic Ethanol: A Joint Research Agenda 〈 http://genomicscience.energy.gov/biofuels/b2bworkshop.shtml〉 (2006)
Food and Agriculture Organization of the United Nations. World Agriculture: Towards 2030/2050 Interim Report 〈 http://www.fao.org/ES/esd/AT2050web.pdf〉 (2006)
McCarthy, E. M. & McDonald, J. F. LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics 19, 362–367 (2003)
Acknowledgements
We acknowledge the contributions of the late M. Gale, who identified the importance of conserved gene order in grass genomes. This work was mainly supported by the US Department of Energy Joint Genome Institute Community Sequencing Program project with J.P.V., D.F.G., T.C.M. and M.W.B., a BBSRC grant to M.W.B., an EU Contract Agronomics grant to M.W.B. and K.F.X.M., and GABI Barlex grant to K.F.X.M. Illumina transcriptome sequencing was supported by a DOE Plant Feedstock Genomics for Bioenergy grant and an Oregon State Agricultural Research Foundation grant to T.C.M.; small RNA research was supported by the DOE Plant Feedstock Genomics for Bioenergy grants to P.J.G. and T.C.M.; annotation was supported by a DOE Plant Feedstocks for Genomics Bioenergy grant to J.P.V. A full list of support and acknowledgements is in the Supplementary Information.
Author Contributions See list of consortium authors below.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Supplementary information
Supplementary Information
This file contains Supplementary Information, Supplementary Tables S1-S18, Supplementary Figures S1-S19 with Legends, Supplementary Acknowledgments and Supplementary References. (PDF 2282 kb)
Supplementary Data
This file shows dot-plot alignments of the sequence of 23 randomly-selected BAC clones compared 2,378,733 finished bp to the whole genome shotgun assembly. The alignment shows only one mismatch in collinearity, demonstrating the accuracy of the final assemblies. (JPG 3992 kb)
Rights and permissions
About this article
Cite this article
The International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768 (2010). https://doi.org/10.1038/nature08747
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature08747
This article is cited by
-
A chromosome level genome assembly of Pseudoroegneria Libanotica reveals a key Kcs gene involves in the cuticular wax elongation for drought resistance
BMC Genomics (2024)
-
Fast-track transformation and genome editing in Brachypodium distachyon
Plant Methods (2023)
-
Genome-wide expansion and reorganization during grass evolution: from 30 Mb chromosomes in rice and Brachypodium to 550 Mb in Avena
BMC Plant Biology (2023)
-
Pangenomic analysis identifies structural variation associated with heat tolerance in pearl millet
Nature Genetics (2023)
-
Discovery of isoflavone phytoalexins in wheat reveals an alternative route to isoflavonoid biosynthesis
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}