
Abstract
The membrane transporter P-glycoprotein, encoded by the ABCB1 gene, influences the pharmacokinetics of anti-cancer drugs. We hypothesized that variants of ABCB1 affect outcome and toxicity in childhood acute lymphoblastic leukemia (ALL). We studied 522 Danish children with ALL, 93% of all those eligible. Risk of relapse was increased 2.9-fold for patients with the 1199GA variant versus 1199GG (P=0.001), and reduced 61% and 40%, respectively, for patients with the 3435CT or 3435TT variants versus 3435CC (overall P=0.02). The degree of bone marrow toxicity during doxorubicin, vincristine and prednisolone induction therapy was more prominent in patients with 3435TT variant versus 3435CT/3435CC (P=0.01/P<0.0001). We observed more liver toxicity after high-dose methotrexate in patients with 3435CC variant versus 3435CT/TT (P=0.03). In conclusion, there is a statistically significant association between ABCB1 polymorphisms, efficacy and toxicity in the treatment of ALL, and ABCB1 1199G>A may be a new possible predictive marker for outcome in childhood ALL.
Similar content being viewed by others

Introduction
The 10-year overall survival rate for childhood acute lymphoblastic leukemia (ALL) is approaching 80% or higher, with many contemporary treatment programs.1, 2, 3 The majority of treatment failures are due to leukemic relapses. However, many of the treatment failures reflect not only the chemo-sensitivity to the different antileukemic drugs used in the protocols but may also depend on inherited single-nucleotide polymorphisms (SNPs) in genes affecting drug metabolism, transport and binding site affinity.4 Owing to the complex combination of chemotherapy in childhood ALL protocols, individual SNPs are unlikely to have measurable effects on drug disposition and cure rates unless they either affect antileukemic agents used extensively in the protocols such as 6-mercaptopurine5 or methotrexate (MTX),6 or when the gene in question affects several anticancer agents, such as the cytochrome P450 family7 or gluthatione S-transferases,8 and potentially the ABCB1 gene.
The membrane transporter P-glycoprotein (P-gp), encoded by the ABCB1 gene, works both as a functional barrier and as an efflux transporter in a variety of tissues, and it can influence the pharmacokinetics of several anti-cancer drugs.9, 10, 11, 12 Variants in the ABCB1 gene have been shown to alter expression and/or function of P-gp.13 Overexpression of P-gp in tumor cells leads to multidrug resistance14, 15, 16 and a number of antileukemic drugs (for example, glucocorticosteroids, anthracyclines and vincristine) are substrates for P-gp. Even though MTX is not regarded as a P-gp substrate, studies of patients in MTX monotherapy showed that the silent ABCB1 polymorphism 3435C>T may affect outcome and toxicity after MTX therapy.17, 18
Studies exploring the clinical impact of ABCB1 SNPs in childhood ALL are few. We have therefore performed a Danish population-based study of the impact of ABCB1 polymorphisms 1199G>A, 1236C>T, 2677G>T/A and 3435C>T on incidence of ALL and risks of relapse and toxicity.
Materials and methods
Patients
Two hundred and forty-six girls and 317 boys, 1.0–14.9 of age (median 4.5 years) were diagnosed with precursor B-cell or T-cell ALL in Denmark from January 1992 to January 2007. Of these, 41 patients were excluded as a result of incomplete genotyping for all polymorphisms due to a lack of DNA material or poor quality of DNA. The remaining 522 patients included in this study, that is, 93% of those eligible during the study period, were treated according to the NOPHO ALL92 (n=307) or ALL2000 (n=215) protocols. Of these, 357 patients were classified as low-risk ALL and 165 as high-risk ALL.3 More than 95% of the patients were Nordic Caucasians. Blood samples from 200 healthy donors; 94 women and 106 men were genotyped to compare ABCB1 variant frequencies for patients and healthy volunteers (Table 1).
Toxicity studies were conducted on 233 children treated at Rigshospitalet, Copenhagen. For the three-drug (doxorubicin, prednisolone and vincristine) induction-therapy toxicity study, all patients with retrievable laboratory data before treatment day 22 were included. To ensure steady-state measuring of MTX, patients were only included in the MTX pharmacokinetic and toxicity studies if end-of-fusion MTX plasma values between 20 and 26 h after the first high-dose MTX (HD-MTX) course were retrievable (n=124).
The Ethics Committee of Copenhagen (j.nr. 01-259108) and the Danish Data Protection Authority (j.nr. 2005-41-4808) approved the study.
Risk grouping and therapy
According to the NOPHO protocol (ALL92 and ALL2000),3, 19 the children were classified as high-risk ALL if at least one of the following parameters were present: white blood cell count >50 × 109 l−1, T-lineage ALL, presence of central nervous system or testicular ALL, translocations t(9;22)(q34;q11) or t(4;11)(q21;q23) (any MLL rearrangement in ALL2000), translocation t(1;19) or hypodiplody (ALL2000 only), the presence of lymphomatous ALL or mediastinal lymphoma, and/or a poor treatment response (>25% blasts in bone-marrow day 15 or >5% blasts in bone-marrow day 29).
During the first 4 weeks of induction therapy, all patients received intrathecal MTX on days 1, 8, 15 and 29, prednisolone (60 mg m−2 per day), weekly vincristine (2.0 mg m−2, maximum 2.0 mg) and doxorubicin (40 mg m−2) on days 1 and 22. In addition, patients with high-risk ALL received an extra dose of doxorubicin on day 8 in the ALL92 protocol. In ALL92 doxorubicin was given as a 24 h infusion, but as a 4-h infusion in ALL2000.3, 19
The post-remission consolidation, re-induction and maintenance therapy phases have previously been described in details.3, 19
HD-MTX:
Children with low-risk ALL (Supplementary Figure S1a) received HD-MTX courses (5 g m−2 per day) three to four times during consolidation at an interval of 14–28 days and five times during maintenance therapy at an interval of 8 weeks. Leucovorin rescue (15 mg m−2) was given from 36 h after start of each HD-MTX course in the ALL92 protocol and from 42 h in the ALL2000 protocol, and was continued at 6-h intervals until plasma-MTX was below 200 nmol l−1.20
Children with high-risk ALL received 8 g m−2 HD-MTX courses two to four times during the consolidation period, with an interval of at least 42 days (Supplementary Figure S1b). The initial leucovorin rescue dose at 36 h was 50 mg m−2 (ALL2000: 15 mg m−2), followed by leucovorin rescue (15 mg m−2) at 6-h intervals until plasma-MTX was below 200 nmol l−1.20 Intrathecal MTX (8–12 mg depending on age) was administered during HD-MTX courses in both low- and high-risk ALL protocols.
Genotyping
The ABCB1 1199G>A (rs2229109), 1236C>T (rs1128503), 2677G>T/A (rs2032582) and 3435C>T (rs1045642) genotypes were determined using pyrosequencing. Genomic DNA was extracted and purified by NaCl-ethanol-precipitation from 1 to 5 ml EDTA-stabilized blood. HotStar-Taq master mixture (VWR International, Stockholm, Sweden) was used for PCR amplification and reactions were carried out on a Mastercycler gradient instrument (Eppendorf, Hamburg, Germany) in a total volume of 25 μl. The SNPs were analyzed by a Pyrosequencing PSQ96MA instrument (Qiagen, Nordic, Sweden) according to the manufacturer’s protocol and as previously described.21, 22 Five hundred and fourteen patients were successful analyzed for all four polymorphisms. The reduced folate carrier polymorphism RFC80G>A in SLC19A1 was analyzed as previously described.6
Pharmacokinetics and toxicity
To quantify the degree of myelosuppression, we used the nadir hemoglobin, platelet and absolute neutrophil counts within the first 3 weeks of induction therapy or within 4 weeks after the first HD-MTX. The maximum plasma alanine aminotransferase level within the first 3 weeks of induction therapy or within 4 weeks after the first HD-MTX was used as marker of liver toxicity. Samples drawn 20–26 h after initiation of HD-MTX were considered to represent plasma MTX steady-state levels.
Statistics
SAS software (version 9.2, SAS Institute, Cary, NY, USA) was used for statistical analysis. Two-sided P-values <0.05 were considered significant. Survival analyses were performed with a basic time scale defined by the date of diagnosis. The duration of event-free survival (EFS) was defined as the time from diagnosis until the date of relapse, death, or the development of a second malignancy (whichever first) or the last known follow-up for event-free survivors. When relapse was considered an event, then death, second malignancy, bone marrow transplantation, protocol failure and changes of protocol were classified as censored events. Patients in first remission were followed until 30 July 2008. Relapse probabilities were estimated using the Kaplan–Meier method. Univariate Cox regression and multivariate Cox regression analysis, stepwise backward selection with stratification by risk group, were used to identify potential risk factors for an event. Model assumptions, including the proportionality assumption, were assessed using Schoenfeld and martingale residual. A general linear model was used for HD-MTX toxicity and pharmacokinetic analyses, and a general linear mixed model with repeated measures was used for induction therapy. In the statistical tests, all data were log transformed. In multivariate analyses, adjustment variables were gender, protocol (ALL92/ALL2000), risk group (high/low) and immunophenotype (pre-B/T). Hazard ratios (HRs) with 95% confidence intervals (CIs) were calculated when appropriate. χ2-test was applied to test for risk of ALL in the ABCB1 polymorphisms. As the ABCB1 polymorphisms are in linkage disequilibrium and thus inter-dependent (Table 2); no correction for multiple testing was done.
Results
No significant association between development of ALL and any of the polymorphisms were found when patients and 200 healthy blood donors were compared (1199G>T/A, n=518, P=0.38; 1236C>T, n=518, P=0.63; 2677G>T/A, n=520, P=0.51; and 3435C>T, n=517, P=0.22). The genotype frequencies in both donor and patient cohorts (Table 1) were in Hardy–Weinberg equilibrium.
In total, 74 patients developed relapse 0.2–8.3 years from diagnosis (median: 2.6 years). Five developed a second malignancy, 22 patients died before first HD-MTX and 5 patients died in first remission.
The 41 patients with 1199GA variant had more than twofold increased risk of relapse (HR: 2.86, 95% CI: 1.52–5.26, P=0.001) compared with the 477 patients with 1199GG variant. No patients had the 1199AA variant. Within the high-risk group (Figure 1a), the 15 patients with 1199GA variant had more than fourfold increased risk of relapse compared with the 149 patients with 1199GG variant in both univariate (relapse: 37, HR: 4.55, 95% CI: 2.08–10, P=0.0001) and multivariate analysis adjusted for protocol, gender and immunophenotype (HR: 4.34, 95% CI: 2.04–9.09, P=0.0001). In contrast, we found no statistically significant differences in risk of relapse in the low-risk group by univariate analysis (relapse: 32, HR: 1.37, 95% CI: 0.42–4.55, n=354, P=0.60) or multivariate analysis (HR: 1.39, 95% CI: 0.42–4.55, P=0.59; Figure 1b), despite that data did not indicate interactions between the genotypes and risk groups. No deaths or second malignancies were registered in the 1199GA patient group compared with 16 deaths and 5 second malignancies in the 1199GG group. The 5-year overall probability of EFS (pEFS5y) for patients with 1199GA variant was 68% (95% CI: 50–80) and for patients with 1199GG variant was 83% (95% CI: 79–86).

ABCB1 polymorphisms and relapse free survival. The Kaplan–Meier survival curve is shown to visualize the difference between 1199GA and AA variants (the only variants found) in high- (a) and low-risk therapy (b), and 3435TT, 3435CT and 3435CC variants (c). The small inserted tables show number of patients in each variant group at specific times. There were 28 (19%) and 9 (60%) events in the high-risk 1199GG and 1199GA variant groups, respectively (a). There were 29 (9%) and 3 (12%) events in the low-risk 1199GG and 1199GA variant group (b). There were 15 (9%), 34 (13%) and 20 (21%) events in the 3435TT, 3435CT and 3435CC variant groups, respectively. There was no difference in relapse-free survival between high- and low-risk group (c).
The 3435C>T polymorphism was significantly associated with risk of relapse (P=0.02). Compared with 96 patients with the 3435CC variant, 421 patients with the 3435TT or 3435CT variants had, respectively, 61% (HR: 0.39, 95% CI: 0.20–0.76, P=0.006) and 40% (HR: 0.60, 95% CI: 0.34–1.03, P=0.06) reduced risks of relapse in multivariate analysis adjusted for risk, immunophenotype, protocol and gender (Figure 1c).
Only one death and no second malignancy were found in the 3435CC variant group compared with eight deaths and four second malignancies in the 3435CT variant group, and six deaths and one second malignancy in the 3435TT variant group. This led to a pEFS5y of 78% (95% CI: 68–86) for the 3435CC patient group and 83% (95% CI: 79–86) for the 3435CT/TT patient group.
In risk group-stratified analyses, no statistically significant differences in relapse risk were found for either the 1236C>T polymorphism (P=0.37) or 2677G>A/T polymorphism (P=0.98). Nor did we find any statistically significant difference in risk of relapse between the ABCB1 haplotypes (Table 3).
The 22 patients harboring both 1199GA and 3435CC variants had almost threefold greater risk of relapse compared with the remaining 402 patients (HR: 2.96, 95% CI: 1.36–6.49, P=0.007).
We looked at interactions between RFC80G>A and both 1199G>A and 3435C>T with respect to risk of relapse, but no statistically significant interaction between 1199G>A and RFC80G>A polymorphisms was found (P=0.14), and interaction between 3435C>T and RFC80G>A was not tested, as none of the 32 patients with RFC80AA and 3435TT had a relapse. Instead, an additive model including RFC80G>A and 3435C>T as covariates was used, and showed that both polymorphisms had effect on outcome (P=0.024). When combining these two polymorphisms, the 78 patients with RFC80GA/GG and 3435CC had an almost fivefold higher risk of relapse (HR: 4.75, 95% CI: 1.70–13.26), and the 207 patients with RFC80GA/GG and 3435CT had almost threefold higher risk of relapse (HR: 2.87, 95% CI: 1.08–7.66) when compared with the 32 patients with both RFC80AA and 3435TT. Multivariate analysis including these polymorphisms, risk group, immunophenotype and protocol gave very similar HRs.
As we have previously demonstrated that SLC19A1 80GA and SLC19A1 80GG had similar effects on outcome,6 another statistical analysis was made, where SLC19A1 variants were grouped in two groups (80AA and 80GA/GG). The ABCB1 1199GA and the 3435CC combined gave the same hazard rate as each independently and were therefore grouped in one group, and the remaining variants, 1199GG, 3435CT and 3435TT, were grouped together. The multivariate Cox regression analysis showed an interaction between polymorphisms in the ABCB1 and SLC19A1 genes (P=0.048). Patients with SLC19A1 80GG/GA and ABCB1 1199GA/3435CC had almost twofold higher risk of relapse as compared with patients with other ABCB1 variants (HR: 1.89, 95% CI: 1.10–3.27). However, patients with SLC19A1 80AA and ABCB1 1199GA/3435CC had more than 10-fold higher risk of relapse than patients with other ABCB1 variants (HR: 10.89, 95% CI: 2.10–56.38). This indicates a synergistic rather than an additive effect of the ABCB1 and SLC19A1 genes, and implies that both genes have an impact on MTX. Thus, MTX could be a substrate for P-gp. This is supported by studies showing impact of ABCB1 3435C>T variants on the toxicity or disease score in MTX monotherapy studies,17, 18 which suggest that ABCB1 variants influence MTX efflux from leukemic cells.
Both univariate and multivariate analyses showed no statistical difference in end-of-infusion MTX plasma levels in relation to any of the ABCB1 variants (P>0.20 in all univariate and multivariate analyses). No pharmacokinetic studies were carried out for doxorubicin, prednisolone and vincristine.
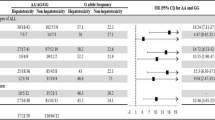
In both uni- and multivariate analyses, ABCB1 polymorphisms significantly influenced the risk of toxicities both after induction therapy and HD-MTX, being most pronounced for 3435C>T (Tables 4 and 5). During induction therapy, patients with the 3435CT or 3435CC variant had significantly less anemia (multivariate P=0.01 and P=0.01, respectively) and less thrombocytopenia (multivariate P=0.0002 and P<0.0001, respectively) when their nadir values were compared with patients with the 3435TT variant (Table 4). Similar effects were seen for neutrophil nadirs when comparing 3435TT patients with patients with the 3435CT variant.
Considering toxicity after HD-MTX, patients with the 3435CC variant had statistically significant higher alanine aminotransferase levels (univariate P=0.03, multivariate P=0.06), when compared with patients with 3435CT or 3435TT variants (Table 5). Including end-of-infusion plasma MTX levels and the RFC80G>A polymorphisms in the models did not significantly alter this effect of 3435C>T.
In the haplotype with strongest linkage disequilibrium 2677GG/1236CC, 2677GT/1236CT and 2677TT/1236TT, we did not see any statistically significant difference in hemoglobin values (P=0.11, multivariate P=0.16), but platelets and neutrophils nadir during induction therapy were statistically significantly lower in patients with 2677TT/1236TT genotype compared with patients with the 1236CC/2677GG genotype (platelets: P=0.01, multivariate P=0.02; neutrophils: P=0.006, multivariate P=0.06).
Discussion
The study underlines the delicate balance between efficacy and toxicity when treating children with ALL with potent chemotherapeutic agents.4 We have previously demonstrated that SNPs, associated with reduced relapse rates, in the reduced folate carrier6 and thiopurine methyltransferase,5 may lead to increased risk of toxicities or even second cancers,23 which ultimately can precipitate in no difference on EFS in a large patient cohort.
To our knowledge, this is the first study to explore the association between ABCB1 1199G>A polymorphism and risk of relapse in childhood ALL. The pronounced increase in relapse rates for high-risk ALL patients with the 1199GA variant is likely to reflect that high-risk patients are more exposed to P-gp substrates (glucocorticosteroids, vincristine, anthracyclines and alkylating agents) than low-risk patients.3 In accordance with these findings, the 1199GG genotype has also been associated with a superior outcome among acute myeloid leukemia patients.24 Furthermore, in vitro studies of HEK and LLC-PK1 cells have showed increased drug resistance to doxorubicin and/or vincristine for the 1199GA variants when compared with the 1199GG variant.25, 26, 27 Lower efflux of vincristine in patients with the 1199GG variants compared with patients with the 1199GA variant may thus explain the effect on relapse. De Meyer et al.28 has found better renal function in kidneys from donors with the 1199GA variant, thus another explanation could be a lower renal function in patients with the 1199GG variant allowing drugs to be retained longer in the organism. Still it remains to be determined whether patients with the unfavorable 1199GA might benefit from higher doses of vincristine or protocols with more emphasis on drugs with a lower affinity for P-gp.
The inferior outcome for ALL patients with the 3435CC genotype found in this study is in agreement with previous publications.29, 30, 31 These clinical observations are supported by in vitro studies of CD56+ cells and the duodenum, which have shown lower P-gp expression and/or P-gp function in 3435TT.32, 33 However, as seen with the 1199G>A polymorphism, the reduced relapse rates for patients with the 3435CT and 3435TT variant were jeopardized by their higher frequency of death in remission and second malignancies.
In agreement with a French study,34 the present study has demonstrated that the ABCB1 1236C>T and 2677G>T/A polymorphisms are in strong linkage disequilibrium, and that both are in moderate linkage disequilibrium with the 3435C>T polymorphism. Accordingly, out of 54 possible haplotypes only 5 haplotypes contained at least 5% of the patients and accounted for >80% of all patients included in the present induction therapy study.
Wang et al.35 found that in spite of the assumed linkage disequilibrium between 1236C>T, 2677G>A/T and 3435C>T, only 3435C>T accounted for altered RNA expression levels. Kimchi-Sarfaty et al.36 have hypothesized that even if the 3435C>T nucleotide substitution does not result in an amino acid change, the C>T substitution may cause an alteration in co-translational folding, which requires a rare tRNA and thereby alters P-gp activity. This is consistent with our finding showing a strong association with the 3435C>T polymorphism and risk of relapse, but not with the 2677G>A/T and the 1236C>T polymorphisms.35
The complex drug combinations applied to cure children with ALL emphasize that multiple genetic variants need to be tested, to understand host-related causes for treatment failure.4, 37 As other studies17, 18 have indicated that MTX could be a substrate for P-gp and SLC19A1 (RFC) encodes for a protein involved in influx of MTX into the cell, and as HD-MTX is widely used in NOPHO protocols,3, 20 we investigated the interactions and, as illustrated in this study, it remains to be clarified whether the additive prognostic effect of variants of the reduced folate carrier (encoded by SLC19A1) and the 3435C>T polymorphism reflects their impact on MTX versus other drugs. However, the hydrophobic substrates vincristine, doxorubicin and prednisolone are thought to enter the cell through diffusion, while the hydrophilic MTX is the only drug entering the cell with help from the reduced folate carrier. In addition, the impact of ABCB1 3435C>T variants on the toxicity or disease score in both the present and in MTX monotherapy studies17, 18 suggest that ABCB1 variants influence MTX efflux from leukemic cells.
The more profound bone-marrow toxicity found in patients with the 3435TT variant after the doxorubicin-containing induction therapy is in accordance with their reduced risk of relapse and increased risk of death or second cancer. Among patients with a favorable prognosis due to the ABCB1 variants they harbor, future studies are needed to explore which additional genetic polymorphisms cause a small subset of these patients to die in remission or develop a second cancer. Such inherited genetic variants may involve glutathione S-transferases, cytochrome P-450 enzymes, quinine oxoreductase, or the folate pathway.38, 39, 40, 41
Although MTX plasma levels after HD-MTX did not differ significantly between subsets of patients defined by their ABCB1 variants, the 3435CC variant was associated with more liver toxicity after the first HD-MTX treatment. In a previous study, we similarly reported higher levels of alanine aminotransferase among the patients with the highest risk of relapse.6 It remains to be clarified whether this reflects intracellular hepatic MTX exposure, a shift in the metabolism in the liver due to the genetic variant or differences in folate disposition or other pathways.
The lack of statistically significant associations between ABCB1 variants and risk of ALL is in agreement with a Hungarian study,42 although other studies with Indian30 and Polish31 patients/controls found higher incidences of ALL among patients with the 3435TT variant. Importantly, the frequency of 3435TT among patients in the Polish study, but not their controls, was very similar to the frequencies found in our and other studies with white individuals, such as the 1000genom project, HapMap studies and the Hungarian study.42 Based on the magnitude of the present study with more than 750 individuals included, the explored ABCB1 variant does not seem to be associated with the risk of ALL.
In the present study, we found that the ABCB1 genetic variants 1199G>A and 3435C>T are associated with outcome in childhood ALL. Overall, >29% of patients with 1199GA variant had relapse, in the high-risk group >60%; thus, patients with 1199G>A polymorphism should be observed more intensively. To further elucidate the role of pharmacogenetics and the biological mechanisms involved, we would recommend pharmacokinetic studies with intracellular drug-level measurements.
This population-based cohort study emphasizes the need to add pharmacogenetic data to the conventional parameters routinely included in survival analyses (for example, leukemia karyotype or post-remission minimal residual disease), to improve prediction of risk in both relapse and toxicity. Further prospective studies are needed to determine whether dose adjustments according to host pharmacogenomics can significantly improve outcome in childhood ALL without unacceptable increments in toxicity.
References
Moricke A, Zimmermann M, Reiter A, Henze G, Schrauder A, Gadner H et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 2010; 24: 265–284.
Pui CH, Pei D, Sandlund JT, Ribeiro RC, Rubnitz JE, Raimondi SC et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24: 371–382.
Schmiegelow K, Forestier E, Hellebostad M, Heyman M, Kristinsson J, Soderhall S et al. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24: 345–354.
Davidsen ML, Dalhoff K, Schmiegelow K . Pharmacogenetics influence treatment efficacy in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol 2008; 30: 831–849.
Schmiegelow K, Forestier E, Kristinsson J, Soderhall S, Vettenranta K, Weinshilboum R et al. Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia 2009; 23: 557–564.
Gregers J, Christensen IJ, Dalhoff K, Lausen B, Schroeder H, Rosthoej S et al. The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood 2010; 115: 4671–4677.
Borst L, Wallerek S, Dalhoff K, Rasmussen KK, Wesenberg F, Wehner PS et al. The impact of CYP3A5*3 on risk and prognosis in childhood acute lymphoblastic leukemia. Eur J Haematol 2011; 86: 477–483.
Borst L, Buchard A, Rosthoj S, Wesolowska A, Wehner PS, Wesenberg F et al. Gene dose effects of GSTM1, GSTT1 and GSTP1 polymorphisms on outcome in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol 2012; 34: 38–42.
Cascorbi I . P-glycoprotein: tissue distribution, substrates, and functional consequences of genetic variations. Handb Exp Pharmacol 2011; 201: 261–283.
Schinkel AH, Wagenaar E, Mol CA, van DL . P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest 1996; 97: 2517–2524.
Terao T, Hisanaga E, Sai Y, Tamai I, Tsuji A . Active secretion of drugs from the small intestinal epithelium in rats by P-glycoprotein functioning as an absorption barrier. J Pharm Pharmacol 1996; 48: 1083–1089.
Zhang Y, Benet LZ . The gut as a barrier to drug absorption: combined role of cytochrome P450 3A and P-glycoprotein. Clin Pharmacokinet 2001; 40: 159–168.
Wang RB, Kuo CL, Lien LL, Lien EJ . Structure-activity relationship: analyses of p-glycoprotein substrates and inhibitors. J Clin Pharm Ther 2003; 28: 203–228.
Biedler JL, Riehm H . Cellular resistance to actinomycin D in Chinese hamster cells in vitro: cross-resistance, radioautographic, and cytogenetic studies. Cancer Res 1970; 30: 1174–1184.
Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM . Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol 1999; 39: 361–398.
Gottesman MM, Pastan I, Ambudkar SV . P-glycoprotein and multidrug resistance. Curr Opin Genet Dev 1996; 6: 610–617.
Kato T, Hamada A, Mori S, Saito H . Genetic polymorphisms in metabolic and cellular transport pathway of methotrexate impact clinical outcome of methotrexate monotherapy in Japanese patients with rheumatoid arthritis. Drug Metab Pharmacokinet 2011; 27: 192–199.
Grabar PB, Rojko S, Logar D, Dolzan V . Genetic determinants of methotrexate treatment in rheumatoid arthritis patients: a study of polymorphisms in the adenosine pathway. Ann Rheum Dis 2010; 69: 931–932.
Gustafsson G, Schmiegelow K, Forestier E, Clausen N, Glomstein A, Jonmundsson G et al. Improving outcome through two decades in childhood ALL in the Nordic countries: the impact of high-dose methotrexate in the reduction of CNS irradiation. Nordic Society of Pediatric Haematology and Oncology (NOPHO). Leukemia 2000; 14: 2267–2275.
Skarby TV, Anderson H, Heldrup J, Kanerva JA, Seidel H, Schmiegelow K . High leucovorin doses during high-dose methotrexate treatment may reduce the cure rate in childhood acute lymphoblastic leukemia. Leukemia 2006; 20: 1955–1962.
Green H, Soderkvist P, Rosenberg P, Horvath G, Peterson C . mdr-1 single nucleotide polymorphisms in ovarian cancer tissue: G2677T/A correlates with response to paclitaxel chemotherapy. Clin Cancer Res 2006; 12: 854–859.
Green H, Soderkvist P, Rosenberg P, Horvath G, Peterson C . ABCB1 G1199A polymorphism and ovarian cancer response to paclitaxel. J Pharm Sci 2008; 97: 2045–2048.
Schmiegelow K, Al-Modhwahi I, Andersen MK, Behrendtz M, Forestier E, Hasle H et al. Methotrexate/6-mercaptopurine maintenance therapy influences the risk of a second malignant neoplasm after childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Blood 2009; 113: 6077–6084.
Green H, Falk IJ, Lotfi K, Paul E, Hermansson M, Rosenquist R et al. Association of ABCB1 polymorphisms with survival and in vitro cytotoxicty in de novo acute myeloid leukemia with normal karyotype. Pharmacogenomics J 2010; 12: 111–118.
Crouthamel MH, Wu D, Yang Z, Ho RJ . A novel MDR1 G1199T variant alters drug resistance and efflux transport activity of P-glycoprotein in recombinant Hek cells. J Pharm Sci 2006; 95: 2767–2777.
Woodahl EL, Yang Z, Bui T, Shen DD, Ho RJ . Multidrug resistance gene G1199A polymorphism alters efflux transport activity of P-glycoprotein. J Pharmacol Exp Ther 2004; 310: 1199–1207.
Woodahl EL, Crouthamel MH, Bui T, Shen DD, Ho RJ . MDR1 (ABCB1) G1199A (Ser400Asn) polymorphism alters transepithelial permeability and sensitivity to anticancer agents. Cancer Chemother Pharmacol 2009; 64: 183–188.
De Meyer M, Haufroid V, Elens L, Fusaro F, Patrono D, De PL et al. Donor age and ABCB1 1199G>A genetic polymorphism are independent factors affecting long-term renal function after kidney transplantation. J Surg Res 2012; 178: 988–995.
Yang YL, Lin DT, Chang SK, Lin SR, Lin SW, Chiou RJ et al. Pharmacogenomic variations in treatment protocols for childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 2010; 54: 206–211.
Rao DN, Anuradha C, Vishnupriya S, Sailaja K, Surekha D, Raghunadharao D et al. Association of an MDR1 gene (C3435T) polymorphism with acute leukemia in India. Asian Pac J Cancer Prev 2010; 11: 1063–1066.
Jamroziak K, Mlynarski W, Balcerczak E, Mistygacz M, Trelinska J, Mirowski M et al. Functional C3435T polymorphism of MDR1 gene: an impact on genetic susceptibility and clinical outcome of childhood acute lymphoblastic leukemia. Eur J Haematol 2004; 72: 314–321.
Drescher S, Schaeffeler E, Hitzl M, Hofmann U, Schwab M, Brinkmann U et al. MDR1 gene polymorphisms and disposition of the P-glycoprotein substrate fexofenadine. Br J Clin Pharmacol 2002; 53: 526–534.
Hoffmeyer S, Burk O, von RO, Arnold HP, Brockmoller J, Johne A et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA 2000; 97: 3473–3478.
Jeannesson E, Albertini L, Siest G, Gomes AM, Ribeiro V, Aslanidis C et al. Determination of ABCB1 polymorphisms and haplotypes frequencies in a French population. Fundam Clin Pharmacol 2007; 21: 411–418.
Wang D, Johnson AD, Papp AC, Kroetz DL, Sadee W . Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet Genomics 2005; 15: 693–704.
Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007; 315: 525–528.
Rocha JC, Cheng C, Liu W, Kishi S, Das S, Cook EH et al. Pharmacogenetics of outcome in children with acute lymphoblastic leukemia. Blood 2005; 105: 4752–4758.
Bolufer P, Collado M, Barragan E, Calasanz MJ, Colomer D, Tormo M et al. Profile of polymorphisms of drug-metabolising enzymes and the risk of therapy-related leukaemia. Br J Haematol 2007; 136: 590–596.
Stanulla M, Dynybil C, Bartels DB, Dordelmann M, Loning L, Claviez A et al. The NQO1 C609T polymorphism is associated with risk of secondary malignant neoplasms after treatment for childhood acute lymphoblastic leukemia: a matched-pair analysis from the ALL-BFM study group. Haematologica 2007; 92: 1581–1582.
Guillem VM, Collado M, Terol MJ, Calasanz MJ, Esteve J, Gonzalez M et al. Role of MTHFR (677, 1298) haplotype in the risk of developing secondary leukemia after treatment of breast cancer and hematological malignancies. Leukemia 2007; 21: 1413–1422.
Woo MH, Shuster JJ, Chen C, Bash RO, Behm FG, Camitta B et al. Glutathione S-transferase genotypes in children who develop treatment-related acute myeloid malignancies. Leukemia 2000; 14: 232–237.
Semsei AF, Erdelyi DJ, Ungvari I, Kamory E, Csokay B, Andrikovics H et al. Association of some rare haplotypes and genotype combinations in the MDR1 gene with childhood acute lymphoblastic leukaemia. Leuk Res 2008; 32: 1214–1220.
Acknowledgements
This study was supported by grants from the Swedish Cancer Society, the Swedish Research Council, the County Concil in Östergötland, the Danish Childhood Cancer Foundation and the University Hospital Rigshospitalet, Denmark. We are thankful for the skilful technical assistance of MSc Ingrid Jakobsen Falk. Likewise, we are thankful to Dr Darrin Bayliss for proofreading the manuscript. We are indebted to all patients for their participation in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website
Supplementary information
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Gregers, J., Gréen, H., Christensen, I. et al. Polymorphisms in the ABCB1 gene and effect on outcome and toxicity in childhood acute lymphoblastic leukemia. Pharmacogenomics J 15, 372–379 (2015). https://doi.org/10.1038/tpj.2014.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2014.81
This article is cited by
-
Non-toxic polymer nanovectors for improved delivery of dexamethasone
Scientific Reports (2021)
-
ABCB1 in dermatology: roles in skin diseases and their treatment
Journal of Molecular Medicine (2021)
-
Impact of ABCB1 Gene (C3435T/A2677G) Polymorphic Sequence Variations on the Outcome of Patients with Chronic Myeloid Leukemia and Acute Lymphoblastic Leukemia in Kashmiri Population: A Case–Control Study
Indian Journal of Hematology and Blood Transfusion (2021)
-
Pharmacogenetics of amfepramone in healthy Mexican subjects reveals potential markers for tailoring pharmacotherapy of obesity: results of a randomised trial
Scientific Reports (2019)
-
Pharmacotherapeutic Management of Wilms Tumor: An Update
Pediatric Drugs (2019)
