
Abstract
We have studied the global frequency distributions of 10 single nucleotide polymorphisms (SNPs) across 132 kb of CYP2C8 and CYP2C9 in ∼2500 individuals representing 45 populations. Five of the SNPs were in noncoding sequences; the other five involved the more common missense variants (four in CYP2C8, one in CYP2C9) that change amino acids in the gene products. One haplotype containing two CYP2C8 coding variants and one CYP2C9 coding variant reaches an average frequency of 10% in Europe; a set of haplotypes with a different CYP2C8 coding variant reaches 17% in Africa. In both cases these haplotypes are found in other regions of the world at <1%. This considerable geographic variation in haplotype frequencies impacts the interpretation of CYP2C8/CYP2C9 association studies, and has pharmacogenomic implications for drug interactions.
Similar content being viewed by others

Introduction
The CYP2C8 and CYP2C9 enzymes are members of the cytochrome-P450 CYP2C subfamily of enzymes that are responsible for metabolizing approximately 20% of pharmaceutical drugs used today. CYP2C8 metabolizes various endogenous compounds including arachidonic acid, retinoic acid, and therapeutic drugs such as the widely used chemotherapeutic agent paclitaxel.1 CYP2C8 and CYP2C9 activities overlap in that both enzymes can metabolize arachidonic acid, several nonsteroidal anti-inflammatory drugs, retinoic acid and others. CYP2C9 separately has been implicated in the metabolism of warfarin, celecoxib, nateglinide and others.2, 3
Both CYP2C8 and CYP2C9, the genes coding for these enzymes, are known to be polymorphic (first reported by Dai4 and Stubbins5), and previously published data suggest frequencies of the variant alleles differ among ethnic populations.6 This genetic variation has been shown to have pharmacological implications. The specific functional consequences of the derived variants depend on the substrate. For CYP2C8 264Met, the activity ranges from 0 to 15% compared to 264Ile,7 and for 269Phe the activity for paclitaxel is 50% that of 269Ile.4 At CYP2C9, activity of 144Cys ranges from 10 to 90% of the activity of 144Arg.2 The derived CYP2C8 variants, 139Lys and 399Arg, which occur together on the same molecule, produce an enzyme with 15–40% of activity compared to the ancestral form,4 depending on substrate. Clearer understanding of the associations can move us a step forward toward individualized drug administration and dosing.8 The two genes are adjacent and within a genomic segment of ∼130 kb on chromosome 10.
Several studies suggest a relationship between CYP2C8 variants, the metabolic index of paclitaxel and drug-related toxicities.4, 9, 10, 11, 12, 13 Though many of those studies showed interindividual variation in paclitaxel pharmacokinetics, some found no association with specific functional variants at CYP2C8, arguing that those variants were not responsible for the variation. However, other variants at CYP2C8 do seem relevant.13 In addition, ethnic distributions of the CYP2C8 polymorphisms suggest that whichever variant alleles affect paclitaxel metabolism may be more common in a particular ethnic group.14, 15, 16 This observation suggests one explanation for varying degrees of toxicities from therapeutic drugs, not limited to paclitaxel, seen in patients of different ethnic ancestries.17, 18, 19, 20, 21, 22 To test the hypothesis that CYP2C8 variation is related to paclitaxel metabolism and toxicity, we are conducting a clinical trial testing the effects of CYP2C8 haplotypes on drug-related toxicities from paclitaxel in different ethnic groups of breast cancer patients. In designing and conducting a genotype–phenotype association study that can also address interethnic genetic variation, an accurate characterization of global variation in target genes is critical. As such data for CYP2C8 haplotypes are scanty, we have undertaken to assemble the necessary dataset. As linkage disequilibrium (LD) between CYP2C8 and CYP2C9 has been reported23 and can be seen in HapMap data, we included some CYP2C9 polymorphisms in this initial study.
Using available public databases detailing known common coding and noncoding polymorphisms, we selected and studied 10 single nucleotide polymorphisms (SNPs) of the CYP2C8 and CYP2C9 genes in populations around the world (Table 1). We also estimated haplotype frequencies for the CYP2C8–CYP2C9 region in these diverse populations. In this paper, we present our initial data demonstrating population-specific allele and haplotype frequencies of CYP2C8 and CYP2C9. This unique dataset will be instrumental in future investigations of differences in CYP2C8-mediated drug metabolism among ethnic populations and in designing clinical trials to examine the putative functional impact of genetic polymorphisms of CYP2C8 and CYP2C9.
Results
CYP2C8-CYP2C9 polymorphism frequencies
Samples from ∼2500 individuals were typed for 10 selected SNPs across a region of 132 kb on chromosome 10 encompassing CYP2C8 and CYP2C9. SNPs across the CYP2C8–CYP2C9 region showed significant allele frequency variation among populations in different geographic regions (Table 1; Figure 1). None of the 450 Hardy–Weinberg tests (45 populations for 10 SNPs) was significant when multiple testing was taken into account. Thus, our assumption that no cryptic variation interfered with the genotyping assays seems reasonable, though we cannot exclude rare sequence variants that may have interfered with the TaqMan assay resulting in a ‘null’ allele. All of the allele frequency data can be found in ALFRED (the ALlele FREquency Database) using the dbSNP rs# in Table 1 as a keyword. The typing of our non-human primate samples unambiguously identified the ancestral human sequences and was in agreement with the data in the UCSC Genome Browser whenever primate sequences were available.

A graphical depiction of the frequencies of the ancestral alleles at the 10 single nucleotide polymorphisms (SNPs) studied in the 45 populations. The figure illustrates the frequencies of these alleles in 45 different populations (listed across the bottom) from around the world. Populations are ordered generally as Africa, Europe, East Asia, Americas.
Haplotype frequencies and linkage disequilibrium
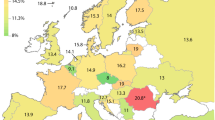
Seventeen common haplotypes were found (Table 2). Estimated haplotype frequencies, plotted in Figure 2, showed that considerable geographic variation exists in haplotype frequencies and that some of the commonly studied functional variants exist on more than one haplotype. Moreover, individual haplotypes contain different combinations of some of the functional variants. Haplotype frequencies are available in ALFRED. There is a complex pattern of pairwise LD that varies among populations as different haplotypes become more or less frequent (data not shown). Because of the large sample sizes studied in many populations, and the large number of populations, we have a clearer understanding of the genetic variation seen previously in individual populations24 and at each locus independently.25 Haplotype G, seen at moderate frequencies (0.05–0.23) only in Europe and Southwest Asia, contains three coding variants: CYP2C8*139Lys, CYP2C8*399Arg and CYP2C9*144Cys. Other combinations of those variants occur in haplotypes H and K. In Africa, the combined frequencies of CYP2C8*269Phe-containing haplotypes B, L and M range from 0.06 to 0.28, but these haplotypes are virtually absent elsewhere in the world. Haplotypes C and N have the CYP2C8*264Ile variant and occur primarily in European populations, but always at low frequencies. Known functional variation is vanishingly rare in Central Asia through East Asia, the Pacific and the Americas.

CYP2C9–CYP2C8 haplotype frequencies. The frequencies of the 17 common haplotypes of CYP2C8–C9 single nucleotide polymorphisms (SNPs). The figure illustrates the frequencies of these haplotypes in 45 different populations listed from top to bottom on the left side in the same order as in Figure 1. The length of each colored bar represents the frequency of the corresponding haplotype in that population. The allelic compositions for the haplotypes are given in Table 2; the frequencies are in ALFRED. Haplotypes that are rare (less than 5%) in all populations are combined into a residual class. Note that coding variants can occur in different combinations and as part of different haplotypes.
Discussion
Genetic variation and cancer therapy
Genetic variation has been considered, for some time, to be a major reason for varying susceptibility of individuals to diseases and responses to drugs. Following the completion of the human genome project, the field of pharmacogenomics has captured the imagination of many clinical investigators with the prospect of realizing personalized medicine. Naturally, cancer pharmacologists have invested significant effort and resources into investigating variation among individuals in drug responses and toxicities to chemotherapy agents, as anticancer agents often have a narrow therapeutic window and consequences of suboptimal dosing can be detrimental. For example, the implications of variant alleles of dihydropyrimidine dehydrogenase on toxicity of 5-fluorouracil-based agents are well described in the literature.26 In addition, polymorphism at CYP2D6 has a predictive value in treatment of breast cancer with tamoxifen, a known substrate of this enzyme. Furthermore, the observation that genetically variant UDP-glucuronosyltransferase 1A1 accounts for significant toxicity from irinotecan therapy led US Food and Drug Administration to recommend dose adjustment according to each patient's genomic profile.
CYP2C8, one of the first human cytochrome P450 genes to be cloned,27 is well recognized as a significant enzyme in metabolism of numerous therapeutic drugs. Considerable interindividual variation in the metabolism of CYP2C8-specific substrates does exist12, 13 and this variation has been associated with polymorphism in CYP2C8, which is reported to be the primary enzyme responsible for the elimination and detoxification of paclitaxel. Several association studies have demonstrated a relationship between CYP2C8 variant alleles, the metabolic index of paclitaxel9, 11, 13, 28, 29 and drug-related toxicities. In addition, the ethnic distributions of these polymorphisms had suggested that variant alleles that affect paclitaxel metabolism are more common in particular ethnic groups.6 Extensive ethnic variation in these polymorphisms is now confirmed in this study, but this study does not directly address the relevance of these polymorphisms to paclitaxel metabolism.
Value of haplotypes
It has become clear that the analysis of a single SNP in a candidate gene may be grossly inadequate in examining genotype–phenotype relationships. Analysis of haplotypes, the combinations of SNP alleles on one chromosome, can show that certain alleles at different SNPs often, or usually, occur on the same chromosomes in the population at frequencies greater than expected by chance, that is, there is LD. Proteins are encoded in cis from each single chromosome; so the combination of coding variants on a chromosome determines the resulting properties of the enzyme encoded by that chromosome. In addition, many cis-acting regulatory sequences can show variation and determine how much of and in which tissues a protein is synthesized. Hence, haplotype analysis is an essential method for detecting associations between sets of genetic variation and gene function whenever more than a single functional SNP may be involved. The value of haplotypes is illustrated by the work of Rodriguiz-Antona et al.13 who studied 12 SNPs across the CYP2C8 gene in a moderate sample of 54 unrelated individuals defined loosely only as ‘Caucasians’. They could not find an effect on paclitaxel metabolism associated with heterozygosity for either the CYP2C8*399Arg and CYP2C8*139Lys containing haplotype or the CYP2C8*264Met containing haplotype. However, a different haplotype, labeled B in their study, did have increased 6α-hydroxylation of paclitaxel. This haplotype B had derived nucleotides in the promoter region, in intron 2 and in intron 7.
Haplotypes in our study
In this study, we genotyped and haplotyped CYP2C8 and CYP2C9 alleles in a large population cohort representing 45 populations from around the world. This dataset represents the most comprehensive study of frequencies of multiple CYP2C8 and CYP2C9 SNPs in diverse populations thus far. We observed striking allele frequency variation of individual polymorphisms among different populations (Figure 1; data in ALFRED). This finding is in line with general expectations for human polymorphisms and long standing clinical observation of ethnic differences in response to and toxicity from various drugs metabolized by the CYP2C subfamily. The global variation in haplotype frequencies (Figure 2; data in ALFRED) similarly shows large interpopulation variation. For example, haplotype G reaches an average frequency of 0.10 in Europe, whereas its frequency averages less than 0.01 in other regions of the world. It is reasonable to hypothesize that some of these population-specific distributions of individual alleles and/or haplotypes may have functional impact, resulting in interindividual and interethnic heterogeneity in drug metabolism, response and toxicity.
Linkage disequilibrium between CYP2C9 and CYP2C8
Three of the coding missense variants are especially interesting from a population genetics perspective and have significant research implications, and potentially clinical implications, in European populations. The three are CYP2C9*144Cys, CYP2C8*399Arg and CYP2C8*139Lys. These three occur in only three haplotypes: G, H and K (Table 2 and Figure 2). Although CYP2C8*399Arg and CYP2C8*139Lys are always seen in cis, they occur with the CYP2C9*144Cys variant in haplotype G and without it in haplotype K. The CYP2C9*144Cys allele occurs by itself in haplotype H. Only in the European and Southwest Asian populations do we see haplotypes G, H and K simultaneously present. In these populations on average 90% of chromosomes with the CYP2C9*144Cys allele also carry these two CYP2C8 variants (Table 3). Thus, an exploratory study in Europeans and/or Southwest Asians of association of the CYP2C9*144Cys allele with a trait (disease risk, therapy response and others) will usually be simultaneously studying CYP2C8 variation. Conversely, on average 89% of chromosomes with either of the two CYP2C8 variants also carry the CYP2C9*144Cys allele. This strong association implies that if any of the three relevant SNPs gives a positive result in a case–control study, it will be difficult to distinguish any association with CYP2C9 from an association with CYP2C8 and vice versa. Given studies of CYP2C8/CYP2C9 pharmacokinetics of ibuprofen,29 it is likely that haplotype effects will be seen for other joint substrates of CYP2C8 and CYP2C9, such as verapamil, isotretinoin, retinoic acid, arachidonic acid and fluvastatin.1, 30 Moreover, given this pattern of LD, there could well be other important variation yet to be identified, especially on haplotype G.
Other functional variation?
Previous studies of variation at CYP2C8 and CYP2C9 may not have uncovered the true wealth of African-specific functional variation. For example, haplotypes B, L and M, containing the CYP2C8*269Phe allele, reach a combined average frequency of 0.17 in Africa, whereas occurring at less than 0.01 in the rest of the world. Thus, it is also clear that 17% of African patients may have a very different drug response, due to their haplotypes with the variant CYP2C8*269Phe allele. In addition, at least three other haplotypes (D, F and O) reach an average frequency of greater than 0.10 in Africa, but are less frequent, on average, in Europe and East Asia, where most previous resequencing efforts have been concentrated. Resequencing a number of African individuals with these haplotypes may discover frequent coding variants, which may provide explanations for pharmacogenomic diversity unexplained by the Arg139Lys, Met264Ile, Ile269Phe and Lys399Arg polymorphisms in CYP2C8 and the Arg144Cys polymorphism in CYP2C9. As we approach the ability to truly examine each individual's DNA sequence to predict optimal therapy, we will need to know what actual genetic variation exists in the population to interpret an individual's DNA sequence.
As a further guide to optimizing design of resequencing efforts to identify additional sequence variation, it is useful to identify the distinct evolutionary sequences that represent lineages that may have independently accumulated variation. To that end we have examined the existing haplotypes we have identified in conjunction with knowledge of the ancestral sequences. Three segments of this 132 kb segment of chromosome 10 show no obligate crossover products. The 17 common haplotypes can be explained by a series of historical crossovers among the different haplotypes of the three segments (Supplementary Material). In addition to testing multiple populations for whatever new polymorphic variation is found, we need to test more existing SNPs in known regulatory regions as well as those that might have less obvious functional consequences: synonymous coding SNPs that could affect mRNA folding and/or mRNA stability as well as intronic SNPs that could affect splicing or may have regulatory function. SNPs recently implicated in paxitaxel pharmacokinetics have especially high priority (for example, 13). Whatever complexity additional SNPs may add to the initial haplotype frequency data we have presented here, additional SNPs can only subdivide the haplotypes defined by the 10 SNPs we have studied and cannot alter the frequencies of the existing combinations of alleles at the missense SNPs.
Lack of comparability across studies
The Rodriguiz-Antona et al. study13 also illustrates a general problem: the need to consider these loci as haplotypes based on a common set of polymorphisms with each haplotype treated as an allele. We are not able to rigorously compare our 8-SNP haplotypes across CYP2C8 (ignoring the 2 SNPs at CYP2C9) with the Rodriguiz-Antona et al.13 12-SNP haplotypes because only four SNPs were studied in common. Based on the four SNPs studied in common, their haplotype B (which shows altered paclitaxel metabolism) falls within a pool of 10 of our 17 common haplotypes (see Supplementary Data). This pool of haplotypes has a total frequency ranging from 68 to 85% among our 10 specific European samples. An obvious future study for us is to add into our dataset the SNPs that specifically identified their haplotype B to allow meaningful comparison of datasets. Similarly, many other studies of CYP2C8 have used only subsets or overlapping sets of polymorphisms to define the relevant haplotypes. We believe that a database of dense marker coverage of each gene in multiple populations will be necessary to define what haplotypes are common in different parts of the world. Such data allow reasonable inference from a few markers of the actual haplotypes present in a future study and allow meaningful comparisons among studies. Our study is an initial step toward such a database.
We believe this study also provides a basis for systematic searches for additional functional variation that may occur at a moderate frequency in one or more regions of the world. When more SNPs are systematically typed across CYP2C8 and CYP2C9 (and the nearby CYP2C18 and CYP2C19), a better picture of the population genetic structure of this region will emerge.
Methods
Markers studied
Using dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), the International HapMap Project (www.hapmap.org), Applied Biosystems' SNPBrowser (http://marketing.appliedbiosystems.com/mk/get/snpb_landing) and the UCSC Genome Browser (http://genome.ucsc.edu/) we compiled data on confirmed SNPs, including the more common functional variants at CYP2C8 and CYP2C9. From this set we selected 10 SNPs likely to be reasonably heterozygous in some part of the world, or confirmed to have functional consequences and frequencies in the polymorphic range, that is, the frequency of the less common allele was >0.01 (Table 1). Of the 10 SNPs, 5 were coding SNPs (cSNPs), 4 in CYP2C8 and 1 in CYP2C9. We emphasized SNPs at CYP2C8 but, based on some reports of LD with CYP2C9,6, 23 we included two SNPs at the 5′ end of CYP2C9. The five noncoding SNPs were chosen to show different frequency patterns among populations already studied in an attempt to maximize haplotype diversity with a small number of SNPs. The final set covered CYP2C8 from just upstream of the coding sequence to just shy of the last exon. The two CYP2C9 SNPs extended the total interval covered to just past the 5′ end of CYP2C9. We did not study variants that had only been reported in single individuals.
Populations studied
We used our resource at Yale University that contains DNA of more than 2500 unrelated individuals representing populations from around the world; this resource has been used for many genetic studies.31, 32, 33, 34 The 45 populations represented in this study include 10 African (Biaka, Mbuti, Yoruba, Ibo, Hausa, Chagga, Masai, Sandawe, Ethiopian Jews and African Americans), 3 Southwest Asian (Yemenite Jews, Druze and Samaritans), 10 European (Adygei, Chuvash, Vologda Russians, Archangel Russians, Ashkenazi Jews, Finns, Hungarians, Danes, Irish and European Americans), 2 Northwest Asian (Komi Zyriane and Khanty), 8 East Asian (Chinese from San Francisco, Taiwan Han Chinese, Hakka, Koreans, Japanese, Ami, Atayal and Cambodians), 1 Northeast Siberian (Yakut), 2 from Pacific Islands (Nasioi Melanesians and Micronesians), 4 North American (Cheyenne, Pima from Arizona, Pima from Mexico, Maya) and 4 South American (Quechua, Ticuna, Rondonia Surui, Karitiana). All subjects gave informed consent under protocols approved by the committees governing human subjects research relevant to each of the population samples. Sample descriptions and sample sizes can be found in ALFRED, the ALlele FREquency Database (http://alfred.med.yale.edu) and in a previous publication.34 We also typed three individuals from each of five other primate groups: Pan troglodytes, Pan paniscus, Gorilla gorilla, Pongo pygmaeus and Hylobates sp.
Marker typing
All markers were typed using TaqMan assays purchased from Applied Biosystems; the assay numbers are given in Table 1. Manufacturer's protocols were followed, with reaction volumes reduced to 3 μl, run in 384-well plates and read on an AB9700HT using Applied Biosystems' SDS (sequence detection system) software.
Statistical analyses
Genotype and allele frequencies for each individual site were estimated by simple gene counting, assuming codominant inheritance with no silent alleles; data were consistent with that assumption. Hardy–Weinberg ratios were tested by χ2 and by an ‘exact’ test using 1000 simulations when small (<5) observed numbers were present for one or more genotype in a population. Haplotypes were estimated using PHASE35 and fastPHASE36 with subpopulation information. There were <1% discrepancies in estimation between the two methods; discrepancies were individually examined. Differences in estimation of missing data for a SNP were responsible for all phasing discrepancies, with the data from PHASE generally being more consistent (fewer rare haplotypes), so the PHASE haplotypes were used. Genotypes of individuals were analyzed by HAPLOT37 for identifying regions with strong LD, sometimes called ‘LD blocks’ (data not shown). We have not attempted to relate these haplotypes to the CYPAlleles nomenclature (www.cypalleles.ki.se) because we have not studied all of the SNPs used in some of the definitions and, as demonstrated here, the allelic definitions for CYP2C9 are not independent of those for CYP2C8.
Conflict of interest
The authors declare no conflict of interest.
References
Totah RA, Rettie AE . Cytochrome P450 2C8: substrates, inhibitors, pharmacogenetics, and clinical relevance. Clin Pharmacol Ther 2005; 77: 341–352.
Kirchheiner J, Tsahuridu M, Jabrane W, Roots I, Brockmoller J . The CYP2C9 polymorphism: from enzyme kinetics to clinical dose recommendations. Per Med 2004; 1: 63–84.
Kirchheiner J, Brockmöller J . Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther 2005; 77: 1–16.
Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI et al. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 2001; 11: 597–607.
Stubbins MJ, Harries LW, Smith G, Tarbit MH, Wolf CR . Genetic analysis of the human cytochrome P450 CYP2C9 locus. Pharmacogenetics 1996; 6: 429–439.
García-Martín E, Martínez C, Ladero JM, Agúndez JA . Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10: 29–40.
Singh R, Ting JG, Pan Y, The LK, Ismail R, Ong CE . Functional role of Ile264 in CYP2C8: mutations affect haem incorporation and catalytic activity. Drug Metab Pharmacokinet 2008; 23: 165–174.
Theken KN, Lee CR . Genetic variation in the cytochrome P450 epoxygenase pathway and cardiovascular disease risk. Pharmacogenomics 2007; 8: 1369–1383.
Mielke S, Sparreboom A, Steinberg SM, Gelderblom H, Unger C, Behringer D et al. Association of Paclitaxel pharmacokinetics with the development of peripheral neuropathy in patients with advanced cancer. Clin Cancer Res 2005; 11: 4843–4850.
Soyama A, Saito Y, Hanioka N, Murayama N, Nakajima O, Katori N et al. Non-synonymous single nucleotide alterations found in the CYP2C8 gene result in reduced in vitro paclitaxel metabolism. Biol Pharm Bull 2001; 24: 1427–1430.
Henningsson A, Marsh S, Loos WJ, Karlsson MO, Garsa A, Mross K et al. Association of CYP2C8, CYP3A4, CYP3A5, and ABCB1 polymorphisms with the pharmacokinetics of paclitaxel. Clin Cancer Res 2005; 11: 8097–8104.
Bahadur N, Leathart JB, Mutch E, Steimel-Crespi D, Dunn SA, Gilissen R et al. CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochem Pharmacol 2002; 64: 1579–1589.
Rodriguiz-Antona C, Niemi M, Backman JT, Kajosaari LI, Neuvonen PJ, Robledo M et al. Characterization of novel CYP2C8 haplotypes and their contribution to paclitaxel and repaglinide metabolism. Pharmacogenomics J 2008; 8: 268–277.
Bravo-Villalta HV, Yamamoto K, Nakamura K, Bayá A, Horiuchi R . A novel intronic mutation that may affect genotyping result of CYP2C8 by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) is strongly associated with CYP2C8*3 in a South American population. J Hum Genet 2007; 52: 195–199.
Daly AK . Pharmacogenetics of the major polymorphic metabolizing enzymes. Fundam Clin Pharmacol 2003; 17: 27–41.
Weise A, Grundler S, Zaumsegel D, Klotzek M, Gröndahl B, Forst T et al. Development and evaluation of a rapid and reliable method for cytochrome P450 2C8 genotyping. Clin Lab 2004; 50: 141–148.
Ohri A, Arena FP . Severe pulmonary complications in African-American patient after bortezomib therapy. Am J Ther 2006; 13: 553–555.
Mattison LK, Fourie J, Desmond RA, Modak A, Saif MW, Diasio RB . Increased prevalence of dihydropyrimidine dehydrogenase deficiency in African-Americans compared with Caucasians. Clin Cancer Res 2006; 12: 5491–5495.
Hasan S, Dinh K, Lombardo F, Kark J . Doxorubicin cardiotoxicity in African Americans. J Natl Med Assoc 2004; 96: 196–199.
Lang CC, Stein CM, Brown RM, Deegan R, Nelson R, He HB et al. Attenuation of isoproterenol-mediated vasodilatation in blacks. N Engl J Med 1995; 333: 155–160.
Hughes LB, Beasley TM, Patel H, Tiwari HK, Morgan SL, Baggott JE et al. Racial or ethnic differences in allele frequencies of single-nucleotide polymorphisms in the methylenetetrahydrofolate reductase gene and their influence on response to methotrexate in rheumatoid arthritis. Ann Rheum Dis 2006; 65: 1213–1218.
Chen ML . Ethnic or racial differences revisited: impact of dosage regimen and dosage form on pharmacokinetics and pharmacodynamics. Clin Pharmacokinet 2006; 45: 957–964.
Yasar U, Lundgren S, Eliasson E, Bennet A, Wiman B, de Faire U et al. Linkage between the CYP2C8 and CYP2C9 genetic polymorphisms. Biochem Biophys Res Commun 2002; 299: 25–28.
Saito Y, Katori N, Soyama A, Nakajima Y, Yoshitani T, Kim SR et al. CYP2C8 haplotype structures and their influence on pharmacokinetics of paclitaxel in a Japanese population. Pharmacogenet Genomics 2007; 17: 461–471.
Makeeva O, Stepanov V, Puzyrev V, Goldstein DB, Grossman I . Global pharmacogenetics: genetic substructure of Eurasian populations and its effect on variants of drug-metabolizing enzymes. Pharmacogenomics 2008; 9: 847–868.
Bosch TM . Pharmacogenomics of drug-metabolizing enzymes and drug transporters in chemotherapy. Methods Mol Biol 2008; 448: 63–76.
Daly AK . Pharmacogenetics of the cytochromes P450. Curr Top Med Chem 2004; 4: 1733–1744.
Scripture CD, Figg WD, Sparreboom A . Peripheral neuropathy induced by Paclitaxel: recent insights and future perspectives. Curr Neuropharmacol 2006; 4: 165–172.
Mielke S . Individualized pharmacotherapy with paclitaxel. Curr Opin Oncol 2007; 19: 586–589.
Zanger UM, Turpeinen M, Klein K, Schwab M . Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem 2008; 392: 1093–1108.
Tishkoff SA, Kidd KK . Implications of biogeography of human populations for ‘race’ and medicine. Nat Genet 2004; 36: S21–S27.
Speed WC, O'Roak BJ, Tárnok Z, Barta C, Pakstis AJ, State MW et al. Haplotype evolution of SLITRK1, a candidate gene for Gilles de la Tourette syndrome. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 463–466.
Mukherjee N, Kidd KK, Pakstis AJ, Speed WC, Li H, Tarnok Z et al. The complex global pattern of genetic variation and linkage disequilibrium at catechol-O-methyltransferase. Mol Psychiatry 2008, Epub ahead of print.
Lee MY, Mukherjee N, Pakstis AJ, Khaliq S, Mohyuddin A, Mehdi SQ et al. Global patterns of variation in allele and haplotype frequencies and linkage disequilibrium across the CYP2E1 gene. Pharmacogenomics J 2008; 8: 349–356.
Stephens M, Scheet P . Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet 2005; 76: 449–462.
Scheet P, Stephens M . A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet 2006; 78: 629–644.
Gu S, Pakstis AJ, Kidd KK . HAPLOT: a graphical comparison of haplotype blocks, tagSNP sets and SNP variation for multiple populations. Bioinformatics 2005; 21: 3938–3939.
Garcia-Martin E, Martinez C, Tabares B, Frias J, Agundez JAG . Interindividual variability in ibuprofeno pharmacokinetics is related to interaction of cytochrome P450 2C8 and 2C9 amino acid polymorphisms. Clin Pharmacol Ther 2004; 76: 119–127.
Acknowledgements
This work was funded in part by NIH grants AA009379 and GM57672 (to KKK). We also acknowledge and thank the following people who helped assemble the samples from the diverse populations: C Barta, FL Black, LL Cavalli-Sforza, K Dumars, J Friedlaender, EL Grigorenko, NJ Karoma, K Kendler, JJ Kim, W Knowler, R-B Lu, A Odunsi, F Okonofua, F Oronsaye, J Parnas, L Peltonen, LO Schulz, Z Tarnok, D Upson, K Weiss and OV Zhukova. Some of the cell lines were obtained from the National Laboratory for the Genetics of Israeli Populations, Tel Aviv University; the African American samples were obtained from the Coriell Institute for Medical Research. Special thanks to the many hundreds of individuals who volunteered to give blood samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
Electronic resources
dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP/
HapMap: http://www.hapmap.org
AB SNPBrowser: http://marketing.appliedbiosystems.com/mk/get/snpb_landing
ALFRED: http://alfred.med.yale.edu/
UCSC Genome Browser: http://genome.ucsc.edu
Supplementary Information accompanies the paper on the Pharmacogenomics Journal website (http://www.nature.com/tpj)
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Speed, W., Kang, S., Tuck, D. et al. Global variation in CYP2C8–CYP2C9 functional haplotypes. Pharmacogenomics J 9, 283–290 (2009). https://doi.org/10.1038/tpj.2009.10
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2009.10
Keywords
This article is cited by
-
The genetic landscape of major drug metabolizing cytochrome P450 genes—an updated analysis of population-scale sequencing data
The Pharmacogenomics Journal (2022)
-
Pharmacokinetics of Roxadustat: A Population Analysis of 2855 Dialysis- and Non-Dialysis-Dependent Patients with Chronic Kidney Disease
Clinical Pharmacokinetics (2021)
-
Investigating selected host and parasite factors potentially impacting upon seasonal malaria chemoprevention in Bama, Burkina Faso
Malaria Journal (2020)
-
Proposed nomenclature for microhaplotypes
Human Genomics (2016)
-
Multi-ethnic distribution of clinically relevant CYP2C genotypes and haplotypes
The Pharmacogenomics Journal (2013)
