
Abstract
MicroRNAs orchestrate brain functioning via interaction with microRNA recognition elements (MRE) on target transcripts. However, the global impact of potential competition on the microRNA pool between coding and non-coding brain transcripts that share MREs with them remains unexplored. Here we report that non-coding pseudogene transcripts carrying MREs (PSG+MRE) often show duplicated origin, evolutionary conservation and higher expression in human temporal lobe neurons than comparable duplicated MRE-deficient pseudogenes (PSG−MRE). PSG+MRE participate in neuronal RNA-induced silencing complexes (RISC), indicating functional involvement. Furthermore, downregulation cell culture experiments validated bidirectional co-regulation of PSG+MRE with MRE-sharing coding transcripts, frequently not their mother genes, and with targeted microRNAs; also, PSG+MRE single-nucleotide polymorphisms associated with schizophrenia, bipolar disorder and autism, suggesting interaction with mental diseases. Our findings indicate functional roles of duplicated PSG+MRE in brain development and cognition, supporting physiological impact of the reciprocal co-regulation of PSG+MRE with MRE-sharing coding transcripts in human brain neurons.
Similar content being viewed by others


Introduction
Brain microRNAs (miRNAs) regulate neuronal transcripts involved in functional pathways,1 and neuronal tissues harbor transcripts with exceptionally long 3′-untranslated regions (3′-UTRs), which are targeted by miRNA regulation.2 Also, evolution of gene-regulating networks3, 4 and regulatory genome components likely modulated brain development and facilitated rapid speciation of humans compared to living primates5 via co-evolution of brain miRNAs and their protein-coding transcript targets.6 The competitive endogenous RNA (ceRNA) hypothesis suggested that non-coding pseudogenes (PSGs) function as decoys for miRNAs, and compete with other transcripts on miRNAs by ’sponging’ reactions. Yet more recently, several studies report active target RNA-directed degradation of miRNAs, rather than passive ‘sponging’ activities.7 Together, this calls for re-inspection of the evolutionary, global in vivo impact, directionality and specificity of the competition between PSGs and protein-coding genes (PCGs) in the human brain.
Most of the protein-coding human genes harbor multiple MREs but non-coding transcripts may be fully devoted to miRNA binding, without interference by active translation.8 Thus, the tumor suppressor gene PTEN, mutations in which are associated with autism, shares MREs with its daughter pseudogene PTENP1, and overexpressing the PTENP1 3′-untranslated region (3′-UTR) led to increased levels of PTEN.9 In contrast, the ‘sponging’ RNA effect (namely, inactivating miRs via facilitating their interactions with transcript targets) is relatively limited for miR-122 targets in hepatocytes,10 and yet other non-coding genes actively induce amplified RNA-directed degradation of miRNAs.11 Furthermore, computational modeling predicted sequential spreading of competition to secondary and tertiary targets,12 compatible with apparent interactions of HMGA2, versican13 and CD44.14 Others argue, mainly with regards to ‘sponging’ processes that such competition depends on miRNA and target concentrations,15 and an in-depth quantitative model predicted a significant competition effect in the mammalian brain.16
We hypothesized that genetic variation in PSGs in schizophrenia17, 18 and autism19, 20 may reflect functional ceRNA regulation and/or target-directed miRNA degradation in the human brain, compatible with the increased trait anxiety and changed miRNA-608 brain targets in human carriers of a single-nucleotide polymorphism (SNP) that disrupts miRNA-608/acetylcholinesterase interactions.21, 22 To approach the global impact of PSGs in the human brain, we explored the evolutionary pattern of MRE-harboring PSGs (PSG+MRE), sought correlations between their expression levels and those of coding mRNAs sharing their MREs in the human temporal lobe and other brain regions and cell types, tested the PSG+MRE/ miRNA/coding gene association in cell culture experiments for reflecting the physiological functions of the MRE-harboring PSG565, and sought interactions of PSG+MRE with several mental diseases of relevant genomic changes.
Materials and methods
Human samples
Two sets of human brain tissues, of varying gyri of the temporal lobe, were received from the Netherland Brain Bank (N=24) and from participants in the Religious Orders Study conducted by investigators at the Rush Alzheimer’s Disease Center, Chicago (N=72). All participants enroll without known dementia and agree to annual detailed clinical evaluation and brain donation. Details of the clinical and pathologic evaluation have been described.23 Subjects were clinically classified as AD dementia (AD), mild cognitive impairment (MCI), and no cognitive impairment (NCI) as described.24, 25 They were pathologically characterized by Braak Stage as described.26 Participants signed an informed consent, an anatomical gift act, and consent to place the data and bio-specimens in a repository for future use. The institutional review boards at the Netherlands Brain bank and the Rush University Medical Center approved these procedures. Frozen material was stored in 1cm slabs in a -80 freezer until pulled and dissected for this study. Full subject data is in Supplementary Tables 3 and 4.
RNA extraction from brain tissue
RNA was extracted with the SPLIT RNA extraction kit (Lexogen, Vienna, Austria) yielding the large RNA fraction with a lower cut-off size of 150 nt. Evaluation on a Bioanalyzer RNA Nano chip (Agilent Technologies) showed medium to high RNA quality (RIN of 6.2 – 8.3). 4 µg RNA was incubated with Terminator 5á-phosphate-dependent exonuclease (Epicentre, Madison, WI, USA) to remove degraded RNA while leaving the capped, full-length mRNA intact.
Reverse transcription and 3′-selective amplification
The complete SQUARE procedure aims at analyzing intact, full-length transcripts (work-flow available on www.lexogen.com). Briefly, 4 μg Terminator-treated RNA was full-length reverse transcribed and extended by a universal 5′ adaptor sequence, and 1/40th of the purified cDNA equivalent to the input of 100 ng total RNA was then amplified in one of 12 parallel PCR reactions. Each PCR reaction was set up with one universal 5′ primer and a 3′ primer selective for one of the 12 possible combinations of the two terminal nucleotides of the mRNA body, immediately upstream of the poly(A) tail. This generated a so-called SQUARE matrix with 12 fields, whereby each of these matrix fields contains the amplification products of an mRNA subpopulation with a given 3′-terminal dinucleotide. Throughout this study, we interrogated all matrix fields combined that correspond to the entire (mRNA) transcriptome. For SOLiD Library preparation and lane mixing, 200 ng of all 288 PCR products (24 RNA samples amplified separately in 12 matrix fields) were heat-fragmented and then ligated to SOLiD-compatible adapters. The libraries were PCR amplified in 17 cycles, with PCR primers indexing each sample with one out of 96 bar codes. Three lane mixes were created, each from 96 libraries corresponding to 24 samples amplified in four matrix fields, dedicating equal molarities to each of the libraries. The three lane mixes were (1) for selective 3′ primer nucleotides AC, AG, CA and GT; (2) AA, AT, CC and CG and (3) CT, GA, GC and GG, and were then further size-selected on a Pippin Prep automated gel elution system (Sage Biosciences, Beverly, MA, USA) in the range of 170-400 base pairs.
For QuantSeq 3′ mRNA-Seq, we used 20 ng of the purified full-length total RNA per sample for performing the 3′ QuantSeq Reverse protocol (Lexogen), resulting in NGS libraries that originate from the 3′ end of polyadenylated RNA. Library generation was started by oligo (dT) priming, with primers already containing the Illumina-compatible linker sequence for Read 1. After first-strand synthesis, the RNA was removed before the second-strand synthesis was initiated by random primers that contained the corresponding Illumina-compatible linker sequence. 3′ QuantSeq generated just one fragment per transcript at the very 3′ end. The libraries were PCR amplified and barcoded in 18 cycles. Equal amounts of the 72 libraries were combined to one lane mixture. Sequencing data are available from NCBI's Gene expression omnibus (GEO), series record GSE70424.
For SOLiD library preparation, lane mixing and RNA sequencing, 200 ng of all 288 PCR products (24 RNA samples amplified separately in 12 matrix fields) were heat-fragmented and then ligated to SOLiD-compatible adapters. The libraries were PCR amplified in 17 cycles, with PCR primers indexing each sample with one out of 96 bar-codes. Three lane mixes were created, each from 96 libraries corresponding to 24 samples amplified in four matrix fields, dedicating equal molarities to each of the libraries. The three lane mixes listed above were subjected to the automated SOLiD EZ Bead System and SOLiD EZ Bead E80 System Consumables (Life Technologies) were applied for template preparations. The SOLiD 5500xl System and single-end chemistry for RNA sequencing was applied (Life Technologies). Sequencing data is available from NCBI's Gene expression omnibus (GEO), series record GSE57152.
For Read alignment, Sequence and quality files, in Lifetech proprietary.xsq binary format, were mapped against the GRGCh38 version of the Homo sapiens genome using the Lifetech Lifescope 2.5.1 whole Transcriptome analysis pipeline. The files produced by this analytical pipeline were coverage; alignment (.bam files); exon junction; gene expression in RPKM with reference to the RefSeq gene structure; and read counts with reference to each gene. Quality control metrics were generated both with the Lifetech suite and with the Integromics SeqSolve analysis suite on all the samples. TopHat 2.0.11 and Cufflinks 2.1.1 suite against GRCh38 genome sequence and associated ENSEMBL exon/transcript annotation in gtf format served to exclude ‘ab initio’ assembled transcripts. Tables of read counts per gene were generated from the alignments using the HTSEQ package. Read lengths were 75-nucleotide fragments, with over 80% genome alignments over the whole sequence length. The minimal sequence base quality value selected for further processing was 10 (Phred score). Bases with a quality value below this parameter were replaced with ‘N’. A progressive alignment method was selected. The minimum genome alignment quality value for an alignment to be processed was again 10 (Phred value). Only primary alignments were considered for gene counts and quantification. The minimal identity seed for alignment extension was 25 nucleotides. The genome mapping percentage of the libraries was always between 60% and 80% of the initial transcripts. Unmapped RNA-seq data (http://www.ebi.ac.uk/gxa/home), which were produced using the same type of sequencing machine as our in-house brain data, were downloaded from this website and processed using the same pipeline as for the brain-derived sequencing data.
For genomic variation tests, human genomic variation in specific genomic sites was calculated using the data of the 1000 genomes project, which includes low coverage genotyping data of more than 2500 individuals of known ethnic origins. (http://www.1000genomes.org/). PSGs were identified by the PseudoPipe algorithm on the most updated genome build GRCh38.
The MicroT algorithm (DIANA lab; http://diana.cslab.ece.ntua.gr/) predicts miRNA-binding sites described previously by Maragkakis et al.26 Briefly, the DIANA-microT 3.0 algorithm consists of (a) alignment of the miRNA sequence on the 3′ untranslated region of a protein coding gene, (b) identification of putative MREs based on specific binding rules, (c) scoring of individual MREs according to their binding type and conservation profile, and (d) calculation of an overall miRNA-target gene score through the weighted sum of all MRE scores lying on the 3′UTR. The algorithm takes into account the known features of miRNA regulation, such as nucleotide composition flanking the binding sites or proximity of one binding site to another within the same 3′UTR.27, 28
RNA extraction from tissue culture
Cells were vortexed for 1 s in 1 ml TRI reagent, followed by incubation for 5 min at room temperature. Two hundred microliters of chloroform was then added, followed by vigorous shaking for 15 s. Next, samples were incubated at room temperature for 5 min, followed by a 15-min centrifugation at 4°C and 12 000 g vortexing. After centrifugation, the aqueous phase was transferred to a new 1.5 ml tube, mixed with 3 × volumes of 100% ethanol (Sigma) and incubated at room temperature for 5 min. Tubes were then centrifuged for 10 min at 4°C and 12 000 g and the liquid discarded. Additional wash was performed with 1 ml 85% ethanol, and precipitates were centrifuged at room temperature and 7500 g. Pellets were then dried and re-suspended in 50 μl nuclease-free water. RNA concentration was measured using Nanodrop-1000 and its integrity assessed by running on a 1% Agarose gel. For long RNA quantification, cDNA was synthesized using the qScript mRNA cDNA synthesis kit (Quanta biosciences, Gaithersburg, MD, USA), and qPCR was performed using Quanta SYBR fastmix (Quanta Biosciences), on a Biorad (Hercules, CA, USA) CFX96 Touch Real-Time PCR Detection System. Expression data were normalized to the geometric average of three housekeeping genes, using the following primers. RPL19: forward - GCTCGATGCCGGAAAAACAC; reverse - GCTGTACCCTTCCGCTTACC; GAPDH: forward - TATAAATTGAGCCCGCAGCC; reverse - TACGACCAAATCCGTTGACTC; ACTB: forward - CCCAAGGCCAACCGCGAGAA; reverse - AGTGGTACGGCCAGAGGCGT. SPSS analysis of the expression data showed normal distribution (KS, P=0.2). For short RNA quantification, cDNA was synthesized using the qScript miRNA cDNA synthesis kit (Quanta biosciences), and qPCR was performed using Quanta SYBR fastmix low ROX (Quanta biosciences), on a Biorad CFX96 Touch Real-Time PCR Detection System. Expression data were normalized to SnoRD-48 (primers were obtained from Quanta biosciences).
For GapmeRs inhibition tests, SHSY-5Y cells were plated on six-well plates. One day later, at a confluence of 80-90%, incorporation of 16-nucleotides-long GapmeR oligonucleotides (one out of three tested from each target gene, Exiqon, Copenhagen, Denmark) was performed using HiPerfect reagent (Qiagen, Venlo, Netherlands) in serum-free medium supplemented with 10% FCS 5 h after transfection. Two hundred microliters of EMEM containing 25 μL HiPerfect reagent and 150 pmol GapmeR was added to each well. Each condition was repeated three times. The following GapmeR sequences were used: –PSG565 - GGCTTAAAAGAGCACT, negative control — AACACGTCTATACGC. PSG565 GapmeR sequence maps uniquely to a transcribed area of the PSG that is not homologous to its parent gene, thereby specifically ensuring avoidance of off-target effects on the parent gene, in addition to general avoidance of off-target effects guaranteed by the manufacturer. Forty-eight hours after transfection, the medium was removed and cells were harvested using 700 μl QIAzol TRI reagent (Qiagen), followed by RNA extraction using miRNAeasy kit (Qiagen) according to kit instructions. RNA concentration was measured using Nanodrop-1000 and its integrity assessed by running on a 1% Agarose gel. DNA was degraded using Ambion DNA-free DNAse treatment and removal reagent (Life Technologies). Consequently, cDNA was synthesized using qScript cDNA synthesis kit (Quanta Biosciences), and qPCR was performed using Quanta SYBR fastmix (Quanta Biosciences), on a Biorad CFX96 Touch Real-Time PCR Detection System. The following primer pairs were used: FOXO3 – forward primer: ATGGGAGCTTGGAATGTGAC; reverse primer: GAGAGCAGATTTGGCAAAGG; PSG565 — forward primer: AAGACTCACATGACCCACCAC; reverse primer: GTACCCTGCAGGTGTGGTCT; SNCA – forward primer: GGCTGAGAAGACCAAAGAGC; reverse primer: ATGACTGGGCACATTGGAAC; STAT3—forward primer: CAGCAGCTTGACACACGGTA; reverse primer: AAACACCAAAGTGGCATGTGA; NeuroD1 — forward primer: GTCCAGCTTGGAGGACCTT; reverse primer: CTGCTCAGGACCTACTAACAACA.
Expression data were tested in SPSS and presented normal distribution (KS, P=0.2).
We used pseudogenes identified by PseudoPipe on the most updated genome build GRCh38. All sequencing data are available from NCBI s Gene expression omnibus (GEO), series records GSE70424 and GSE57152. Code is available upon request from barbashshahar@gmail.com.
Results
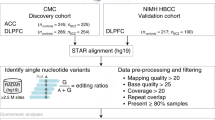
To address the competition between PSGs and coding genes in the human brain, we measured global genomic variation and signals of selective evolutionary sweep using the human 1000Genomes project and animal genome data sets, searched genome-wide association studies data sets for PSGs association with several mental diseases (Figure 1a), and sequenced coding and non-coding polyadenylated RNA, including PSGs in 96 specimens of the human temporal lobe; a brain region implicated in mental diseases such as schizophrenia, autism and bipolar disorder29, 30 (Figure 1b, see Online Methods for details on the cohorts and processing approaches). We also interrogated publicly available sequencing data sets from the human cingulate gyrus and hippocampus and additional, neuronal and non-neuronal tissues, as well as temporal lobe single-cell sequencing and expression data sets (Figure 1b). Expression differences between comparable subsets of PSG+MRE and duplicated PSG−MRE enabled cross-comparison with the expression levels of the corresponding miRNAs and the coding RNA transcripts carrying shared MREs with them. Notably, these were often not their mother genes. Finally, we performed over- and under-expression validation experiments involving selected PSG-coding genes-miRNA-coding transcript targets, in neuroblastoma cells.

Study design at the genomic and expression levels. (a) Human and chimpanzee genomic data were analyzed to extract genomic variation around specific genomic regions and to calculate deviation from neutral evolution. Data of single-nucleotide polymorphism (SNP) association with mental diseases were downloaded from the Psychiatric Genomics Consortium and analyzed for enrichment around pseudogenes (PSGs). (b) Sequencing data from two cohorts of human temporal lobe samples was generated with two methods of library construction for RNA sequencing and two sequencing machines and analyzed for differential expression and co-expression of potential RNA regulators with their targets. Similar analyses were performed for hippocampus and cingulate gyrus based on the data downloaded from the Stanley Neuropathology Consortium. Cell specific RNA-seq data was downloaded from33 and analyzed for differential expression between different PSGs. Regions expressed and incorporated into the Ago2: miRNA complex were downloaded from1 and analyzed for different representations of PSG subsets. Expression data for other, non-neuronal tissues were downloaded from the Tissue Expression Atlas and analyzed for differential expression between different PSG subsets. GapmeR interference tests suppressed either PSGs or protein-coding genes (PCGs) and tested the outcome on the corresponding miRNAs and their PCG and PSG targets. (c) Duplicated PSGs are more likely to harbor MREs. Shown are stacked bar graphs of the percentages of duplicated, processed and ambiguous PSGs in the entire PSG set, Bootstrap PSG and the subsets of PSG+MRE and PSG−MRE. (d) PSG+MRE are more similar to their parent genes than other PSGs. Shown are cumulative distribution functions of the % of similarity between all duplicated PSGs and their parent genes, for PSG+MRE (red, 54 duplicated PSG+MRE), duplicated PSG−MRE and 1 STD around the mean for Bootstrap PSG (blue, 2460 PSGs; Kolmogorov-Smirnov test P<0.01). (e, f) Global conservation of PSG+MRE is particularly apparent at the transcription end sites. Shown is SNP density as a function of genomic location, smoothed with an averaging sliding window of 10%, for PSG+MRE (red line) and duplicated PSG−MRE (blue line) compared to bootstrap analysis (green line), centered on PSG transcription start sites, and averaged for different human sub-populations (Kolmogorov-Smirnov test P<0.001, b; P<0.0001). (g) Increased genomic conservation and evidence of selective sweep around PSG+MRE. Fay and Wu H test of neutrality around PSG+MRE shows decreased values in Yuroba sub-populations compared to PSG−MRE and 1 STD around the mean for Bootstrap PSG (see Supplementary Figure 1 for other sup-populations).
PSG+MRE show evolutionary conservation in human populations
The human genome includes 16 903 annotated PSGs (Ensembl and NCBI genome browsers), but only 123 of the PSGs in these search tools (1%) harbor at least one predicted MRE with a high prediction score (<−15 (microT DIANA lab prediction algorithm). In comparison, we found 28.16% MRE-carrying PCGs (6502 out of 23,091 based on Ensembl annotation). In evolutionary terms, PSGs could be globally sub-classified into duplicated, processed and ambiguous origins (www.pseudogene.org; 15, 55 and 30%, correspondingly, out of the 16 903 annotated PSGs). Duplicated PSGs arise as a result of gene duplication events and include non-exonic regions, whereas processed PSGs have largely emerged by retrotransposition events and are exclusively composed of exons, including Poly A tails.31 Intriguingly, PSG+MRE include 60, 25 and 15% fractions of duplicated, processed and ambiguous PSGs (Figure 1c), a 4-fold higher prevalence of duplicated PSG+MRE compared with PSG−MRE, total PSGs and Bootstrap results (P<0.001; Fisher exact test). Also, PSG+MRE have shown higher similarity with their parent genes compared to PSG−MRE (Figure 1d), indicating more recent divergence from them.
Next, we compared PSG+MRE and duplicated PSG−MRE, two groups whose length, composition and expression levels are close to each other. We employed the 1000Genomes project’s data to compare the evolutionary conservation of PSG+MRE to duplicated PSG−MRE by calculating SNP density in sliding 400 nucleotide windows centered at genes’ start and end sites. PSG+MRE showed tighter conservation, with similar strength for different human sub-populations compared to duplicated PSG−MRE, both in their promoter regions (TSS, 150 nucleotides upstream to the start sites, Figure 1e) and in their end site regions that are usually denser with MREs (TES, 100 nucleotides upstream to the end sites, Figure 1f; Supplementary Figure 1a and b). Using Bootstrap analysis, with 10 000 permutations, for total PSGs, we randomly chose 123 genes out of the entire PSGs list, calculated their averaged genetic variation and s.e.m. and compared it to PSG+MRE. Bootstrap distribution of total PSGs was significantly different from that of PSG+MRE in both examined regions (Supplementary Figure 1c, d), excluding the structural composition of PSG+MRE as a cause for their observed evolutionary distinction.
We further employed the PhyloP algorithm to calculate P-values for deviation from neutral conservation across groups of organisms (Online Methods; list of organisms in Supplementary Table 1). Non-human mammals, vertebrates and primates, including chimpanzee did not show conservational differences between PSG+MRE and PSG−MRE at both the start and end site regions (Supplementary Figure 1e–g). However, TSS and TES values of PSG+MRE and duplicated PSG−MREs in humans reaffirmed the differences we discovered, potentially suggesting that PSG+MRE acquired recent conservation during human speciation. Next, we searched for deviation from neutral evolution by calculating Fay and Wu H test values for windows of 200 nucleotides centered around the identified sites of decreased variation (Figure 1e and f) in 1000Genomes human populations, for 100 nucleotides upstream of transcription start site and 100 nucleotides upstream of transcription end site. The YRI (Yuroba) subpopulation presented significantly lower H values around PSG+MRE transcription start sites, compared to both PSG−MRE and Bootstrap distribution, suggestive of a recent selective sweep (or many small sweeps) in this region (Figure 1g). YRI transcription end and start sites in other sub-populations showed statistically insignificant effects (Supplementary Figure 2a–c). Also, applying similar tests to the genetic variations of expressed and non-expressed PSG−MRE revealed a statistically insignificant trend of lower genetic variation around expressed PSG−MRE (Supplementary Figure 3a–e). The observed evolutionary conservation of PSG+MRE was hence unlikely to simply reflect their structural composition and length.
PSG+MRE show increased expression in human brain neurons
To directly examine PSG+MRE expression, we extracted total RNA from two separate sets of adult human temporal lobe samples (full sample information in Supplementary Tables 2 and 3). Most PSGs predictably showed much lower expression levels than protein-coding genes (Figure 2a), but some polyadenylated PSG+MRE have consistently shown higher expression levels in the temporal lobe than PSG−MRE and Bootstrap distribution, in both cohorts and sequencing methods (Figure 2b and c), and both when averaged across samples and in each individual in the cohort separately (Supplementary Figure 4a). Comparing polyadenylated PSG+MRE to polyadenylated PSG−MRE transcripts predicts duplicated origins for the tested PSG+MRE. PSG−MRE and PSG+MRE showed 8.4 and 52.9 expression levels measured in fragments per kilobase of exon per million reads mapped (FPKM), respectively, in the 72 samples cohort of brain temporal lobe tissues. The parent genes of these PSG+MRE and PSG−MRE showed similar expression levels (Supplementary Figure 4b), excluding the option that aligned PSG+MRE reads originated from the parent genes.

Duplicated PSG+MRE show higher expression levels than PSG−MRE in neuronal tissues. (a) Globally, pseudogenes (PSGs) are less efficiently expressed in the brain than protein-coding genes. Shown is a box plot of total read FPKM values of protein-coding genes and PSGs in the human temporal lobe. (b) Polyadenylated transcripts of PSG+MRE genes are more efficiently expressed in the human temporal lobe than PSG−MRE. Shown are cumulative distribution functions of expressed PSG+MRE (red), PSG−MRE (Blue) and 1 STD around the mean for Bootstrap PSG (KS test P-value= 5.8 × 10−10) for the first human cohort (n=24), library preparation with SQUARE protocol and SOLiD sequencing. (c) Same as (b) for a second human cohort (n=72), library preparation with 3`QuantSeq and Illumina sequencing. (d) –log2 of the KS P-value and fold change of increased expression of PSG+MRE compared to PSG−MRE in the noted human neuronal and non-neuronal tissues. (e) Sequences of the PCR products of HLA-C and its MRE-sharing PSG+MRE; aligned according to the Sanger sequencing plots. Note that at points of variations between the parent gene and this PSG+MRE, the PCR product presents the HLA-C variation exclusively (arrows). (f) Same as e for the HLA-C PSG+MRE PCR product. Note that at points of variations between the parent gene and this PSG+MRE, the PCR product presents the PSG variation exclusively (arrows). (g) PSG+MRE genes are more efficiently expressed than PSG−MRE and Bootstrap PSG in neurons, but not astrocytes and oligodendroglia dissected from human temporal lobes.
Similar to analyses on RNA sequences from the temporal lobe, cingulate gyrus and hippocampus samples from the Stanley Neuropathology Consortium, our in-house data sets of human leukocytes32 and 32 additional tissues using the expression atlas RNA-seq data (http://www.ebi.ac.uk/gxa/home) revealed that the brain presented the most significant expression differences between PSG+MRE and PSG−MRE (-Log2 P-value ~30 compared to ~10 in liver, leukocytes and heart; Figure 2d and 32 additional non-neuronal tissues in Supplementary Figure 4c, Supplementary Table 4). In brain-expressed PSG-targeting miRNA data downloaded from MicroRNA.org (http://www.microrna.org/microrna/home.do), 96, 92 and 95% of the expressed miRNAs emerged as potentially targeting at least one PSG in the human hippocampus, cerebellum and midbrain (Supplementary Figure 4d), with hippocampal miRNAs prevailing. In addition, we found no DNaseI hypersensitivity spots in the genomic domains surrounding PSG+MRE, excluding global genomic differences in their regulation (Supplementary Figure 4e). To test if we correctly avoided identifying parent sequencing reads as PSGs, we designed two primer sets of the histocompatibility gene HLA-C whose 3′ ends target points of variation between HLA-C and its PSG. Both the HLA-C and its PSG PCR products showed complete sequence specificity (Figure 2e and f), and the HLA-C and its PSG product showed similar expression levels (Supplementary Figure 4f). To exclude higher transcriptome depth in brain tissues as an origin of these differences, we analyzed this depth across the available tissues and found no change (Supplementary Figure 4g-i). We conclude that PSG+MRE are expressed at higher levels than PSG−MRE in the human brain, more than in other tissues.
Next, we analyzed cell-type-specific RNA sequencing data sets including 78 postnatal neurons, 32 astrocytes and 22 oligodendroglia cells based on cell-type-specific markers (Supplementary Figure 5a, b).33 Neurons, but not astrocytes and oligodendroglia presented significantly higher expression of PSG+MRE compared to PSG−MRE and to Bootstrap distribution (Figure 2g). In data sets of cell-type-specific transcripts,33 both of duplicated PSG+MRE and of those coding transcripts that share MREs with them presented correlated expression levels in healthy brain tissues and also between Alzheimer’s disease and healthy control tissues. The observed differences were hence unlikely to derive from cell composition changes.
To study the origin of the unique expression patterns of PSG+MRE in neuronal tissues we next analyzed data sets of histone modifications with known effects on transcription. The Roadmap Epigenomics Project’s chromatin immunoprecipitation Chip-Seq data cover the transcription repressor H3K27me3 and the transcription activator H3K4me3ref. [34] in the human temporal lobe. Interestingly, the regions surrounding PSG+MRE showed lower levels than those surrounding PSG−MRE of the suppressor H3K27me3 modification, (comparable to that of highly expressed coding genes; Figure 3a). In both protein-coding genes and PSG+MRE, these troughs of H3K27me3 signals overlapped with the transcription start site or were shifted by several tens of nucleotides from it (see Figure 3b for two examples of each case). In contradistinction, the genomic regions surrounding PSG+MRE and PSG−MRE showed similar levels of the activating H3K4me3 modification (Figure 3c). Taken together, these results indicate that the lower epigenetic repression of PSG+MRE compared to PSG−MRE may operate as a driving force for their more potent expression in human brain neurons.

PSG+MRE show lower suppression-associated histone modifications than PSG–MRE and present distinct microRNA recognition elements (MREs) from their mother genes. (a) Relative enrichment of the transcriptional suppression-associated H3K27me3 histone modification from human temporal lobe around highly (H), moderately (M) and lowly (L) expressed coding genes, as well as around PSG+MRE and PSG–MRE, as a function of genomic location centered at the transcription start site (TSS). (b) Examples of the H3K27me3 enrichment pattern for two coding genes (PLOD1, MRTO4) and the two noted PSGs. (c) Same as a for the transcription activation-associated H3K4me3 histone modification. (d) Limited MRE overlap between brain-expressed PSG+MRE and their parent genes. Three options of evolution changes in the MREs of PSG+MRE: gain, loss or conservation. (e) Numbers of analyzed PSGs. (f) Number of events of more MREs in parent genes compared to PSG+MRE (purple) and vice versa (magenta). (g) A side view of Venn diagrams for MRE overlaps between PSG+MRE and their parent genes. Inset in the lower half: cartoon showing shared MREs on a parent gene and its daughter PSG. All resultant Venn diagrams are centered on the y axis around the margin of the parent gene’s circle. Values on the upper and lower parts of the Y axes indicate MRE numbers on the PSGs and overlap with the parent genes, respectively. 'N' near PSGs denotes ‘Neuronal’ origin (that is, PSGs expressed in the temporal lobe data sets). For the genes ‘GSTA3’ and ‘BMS1’, a front view of the Venn diagram is shown. PSGs, pseudogenes.
PSG+MRE, miRNAs and MRE-sharing coding genes show inter-related regulation
The 16 903 PSGs included in the human genome emerged from 5051 PCGs (some of which evolved into more than one PSG). We surmised that during primate evolution, these PSGs could lose or keep the MREs of their parent genes, or acquire new MREs (Figure 3d). Of the 123 PSG+MRE, we found 57 that belong to pairs of parent-daughter PCG-PSG genes carrying MREs in both partner genes (microT prediction algorithm), allowing calculation of MRE overlaps (Figure 3e). Surprisingly, most of these paired PSGs carried less MRE than their parent genes (Figure 3f), with limited overlap of MREs with their parent genes (Figure 3g; for zoom-in on these genes see Supplementary Figure 6a), equal to that predicted by 10 000 random permutation tests across PSGs and PCGs, across the human genome (Supplementary Figure 6b). However, both the principal ceRNA hypothesis and the more recent concept of target-mediated destruction of miRNAs merely require the existence of shared MREs in a PSG and any expressed coding gene, not necessarily its parent gene. Therefore, we sought potential regulatory interactions in the temporal lobe between the global repertoire of PSGs, MRE-sharing PCGs and other PSGs across the entire genome.
To find whether PSG+MRE participate in the RNA-induced silencing complex (RISC), we analyzed Ago2 HITS-CLIP data of frontal cortex transcripts that were precipitated together with miRNAs using Ago2 antibodies.1 There were approimately six times more coding genes carrying Ago2 clusters on average than PSG-carrying ones in the precipitated complexes. Clusters including PSG+MRE were approximately sixfold-enriched compared to PSG−MRE-including ones, as well as to Bootstrap PSG-included ones, and the PSG+MRE/PSG−MRE expression ratio was 6.3 (Figure 4a). This supported the prediction that PSG+MRE actively participate in Ago2-mediated miRNA regulation, with similarly effective selection of PSG+MRE and coding transcripts onto the RISC complexes.

PSG+MRE associate with RISC complexes and show global regulatory gene pair interactions; not necessarily with their parent genes. (a) Mean and SEM of Ago2:miRNA clusters including PSG and coding gene transcripts show sixfold more identified Ago2:miRNA clusters including PSG+MRE than PSG–MRE or Bootstrap PSG (1-Way-ANOVA P<0.05 and post hoc Tukey's test P<0.05). (b) Schematic expression correlation matrix calculated for protein-coding genes (PCGs) with themselves, or with all PSGs and for PSGs with themselves. Gene pairs with significant correlation coefficients >0.7 were extracted, and the % overlap in MRE was calculated between them as the number of shared MREs divided by the sum of MREs in both genes. (c) Histogram of the % MRE overlap values of the gene pairs with significant correlation coefficient>0.7, for 10 000 permutations, and for the un-permuted data (red line) between PCGs and themselves, PSGs and PCGs and PSGs and themselves. Data based on the first human cohort (n=24), library preparation with SQUARE protocol and SOLiD sequencing. (d) Same as c for the second human cohort (n=72), library preparation with 3`QuantSeq and Illumina sequencing. Note scheme below, reflecting active ceRNA effect between PSG and PCG and among PSGs sharing MREs, but not PCGs. ANOVA, analysis of variance; MREs, microRNA recognition elements; PSGs, pseudogenes.
An expression correlation matrix across 24 and 72 temporal lobe samples (first and second cohort) for all MRE-harboring genes, detected those gene pairs that present significant expression correlation (with correlation coefficient >0.7) across the examined cohorts, and averaged the % MRE overlap between them (Figure 4b; see Supplementary Figure 6c, d for example correlation matrices). Randomly permuting across gene−MRE matched sequences, 10,000 times, allowed calculating permutation P-value to attribute statistical significance. This analysis showed a significant effect for PSG+MRE with global PCGs in both the first and second cohorts (Figure 4c and d left; 1197 gene pairs, permutation calculated P-value=0.012 and 0.045, respectively) and a yet tighter effect among the PSG+MRE with each other (Figure 4c and d, center; 1034 gene pairs, permutation calculated P-value=0.001 and 0.06, respectively). In comparison, we found a non-significant effect among PCGs by themselves (Figure 4c and d right, 12 951 gene pairs), whereas adipose tissue (GSE16615, 39 samples) and peripheral whole blood (GSE22229, 58 samples) showed no effect (Supplementary Figure 7a-c). These findings, observed in two independent human cohorts, two distinct library construction methods and two sequencing machines, suggest that within the temporal lobe, PSG+MRE contribute to mutual RNA regulation by shared MREs in non- parent-daughter gene pairs more effectively than PCGs.8
GapmeR suppression indicates PSG+MRE-PCG-miRNA interactions
To mimic the physiological and cellular aspects of PSG+MRE function in brain neurons, in an in-depth manner, we performed an exemplary GapmeR knockdown experiment in the human-originated SHSY-5Y neuroblastoma cells targeting a highly expressed pseudogene, PGOHUM00000243565, hereby labeled PSG565 for brevity. PSG-565 is particularly suitable for these suppression tests since it shares with regulatory coding transcripts MREs for several miRNAs that are highly expressed and relevant in brain functions, including miR-7-5p, miR-124-3p and miR-23a-3p. PSG-565 knockdown (by 75%, Supplementary Figure 8) caused significant upregulation of its targeting miR-7-5p, supporting target RNA-directed miRNA degradation11 rather than merely a sponging effect, where no increase would be expected. This miR-7-5p increase was predictably accompanied by significant downregulation of its targets alpha Synuclein and the circular RNA CDR1-AS,35 supporting functional relevance. In comparison, miR-23a-3p only showed a trend for upregulation, and its FOXO3 target inversely showed a trend of downregulation, whereas the highly expressed miR-124-3p and its STAT3 and NeuroD1 targets were largely unaffected (Figure 5a; statistics included). The observed triple PSG-miRNA-PCG interactions may operate via shared MREs for PSG-565 in its physiological context, and differentially affects at least six coding genes that map to three different chromosomes and play seminal roles in brain function (Figure 5b).

Suppression of PSG-565 differentially increases the levels of its targeting miRNAs and suppresses protein-coding genes (PCGs) targeted by these miRNAs, indicating functional interactions. (a) SHSY-5Y cells were exposed to antisense GapmeRs inhibiting PSG565. The levels of predicted miRNAs and their PCG targets as measured by quantitative PCR (qPCR) are shown. Data boxes for inhibition groups are dark green and for control groups are purple. MiR-7 was elevated whereas its targets SNCA and CDR1-as were downregulated; miR-23a-3p showed a trend for downregulation and so did its target FOXO3, and the highly expressed miR-124-3p, as well as its targets STAT3 and NeuroD1 were unaffected (statistics for Student's t-test appear in the Figure), supporting the notion that downregulated PSG levels lead to increased levels of cellular miRNAs targeting these PSGs, potentiating the suppression of MRE-sharing PCGs. N=3. (b) Shown are the chromosomal locations of the examined PSG-565 and coding genes. PSG-565 and PCGs with depicted ceRNA functions map to several chromosomes, indicating a wide genomic effect. (c) PSG+MRE are enriched in single-nucleotide polymorphisms (SNPs) associated with mental diseases. Shown are cumulative Distribution Functions (CDFs) for P-values of the association of total SNPs in PSG+MRE, PSG−MRE and PCGs (total SNP numbers 243,785, 240,899 and 241,998) with the noted three mental diseases, at a region of 0-0.1 associations. Autism, bipolar disorder and schizophrenia included 2130, 2103 and 2108 SNP subsets in the regions surrounding PSG+MRE, respectively. (d) Neurotransmission associated SNPs. Shown are CDFs of P-values of the association of SNPs in PSGs that share (red) or do not share (blue) MREs with neurotransmission- involved genes (summated glutamatergic, GABAergic and cholinergic-related genes). Asterisks denote KS P-value <0.05. (e) Scatter plot showing expression correlation between HLA-C and the 8 MRE-sharing PSG-565 (correlation coefficient and P-value also shown on the graph). MRE, microRNA recognition elements; PSGs, pseudogenes.
PSG+MRE carry SNPs associated with mental diseases
To test for functional relevance of brain PSG+MRE, we sought potential association of known SNPs in PSG+MRE with Alzheimer’s disease, autism,36 bipolar disorder,37 schizophrenia,38 ADHD39 and major depression disorder40 (http://www.med.unc.edu/pgc/downloads), all considered to involve many genes.41 We scanned windows of 500 nucleotides upstream and downstream of the transcription start and end sites of PSG+MRE, PSG−MRE and other genomic regions and addressed the cumulative effect of many weakly associated SNPs located around a genomic region (e.g. SNP association enrichment42, 43). Disease-associated SNPs emerged in PSG+MRE with autism, schizophrenia and Bipolar disorder compared to P-values of SNPs in the global genome or around PSG−MRE and PCGs (Figure 5c and Supplementary Figure 9a and b). This was confirmed by Bootstrap analysis (Supplementary Figure 10a), for both the regions flanking the transcription start and end sites and the transcribed regions in PSG+MRE (Supplementary Figure 10b). No enrichment was detected in Alzheimer's disease SNP association data (downloaded from the Alzheimer Disease Neuroimaging Initiative; http://adni.loni.usc.edu/), depression or ADHD. Notably, PSG+MRE sharing MREs with neurotransmission-involved PCGs (65 PSG+MRE, based on KEGG; http://www.genome.jp/kegg/) were more enriched with disease-associated SNPs than other PSG+MRE (Figure 5d).
To test those SNPs that belong to expression-related quantitative trait loci (eQTL) that associate with brain differential expression (the QTL browser, http://www.bios.unc.edu/research/genomic_software/seeQTL/), we separately examined 3679 ‘cis-eQTLs’ localized in chromosomal regions of differentially expressed genes and 37 806 ‘trans-eQTLs’ localized far away from differentially expressed genes. Of those, 19 ‘cis-eQTLs’ and 44 ‘trans-eQTLs’ were identified in windows of 500 nucleotides upstream and downstream of the transcription start and end sites of PSG+MRE. ‘Cis-eQTLs’ showed enrichment for association with autism and schizophrenia and ‘trans-eQTLs’ showed enriched association with Bipolar disorder and Schizophrenia (Supplementary Figure 10c). Tests of robustness involved changing the microT score cut-off of -15 for MRE on PSGs for −15, −20 and −25, and yielded robust results for the identified PSG+MRE effects (see Supplementary Figure 11a–h for test of robustness for different microT score cutoffs for all the reported measurements). Also, comparing the KS statistics to Wilcokson rank sum and to Kruskal–Wallis (Mann–Whitney) analyses for the second data set (n=72) RNA-seq, where the effect was relatively small, both came up with a P-value of 2.8 × 10−17, considerably more significant than the KS P-value of 1.2 × 10−8.
One example of a PCG / PSG interaction that might be linked in a competition and/or target-mediated miRNA destruction manner involves the HLA-C gene, implicated in schizophrenia,44 and PSG-565, which is an HLA-C-unrelated PSG and served for our experimental validation tests. HLA-C and PSG-565 show no parental correlation, yet share 8 MREs among themselves and with other PCGs, e.g. ACHE (Supplementary Table 5). Correspondingly, HLA-C and PSG-565 showed an expression correlation coefficient of 0.76 and a correlation P-value of 0.00001 in temporal lobe tissues (Figure 5e). Given our exemplary experimental interference test (Figure 5), these findings further predict HLA-C related changes in neurotransmission and are indicative of a physiological role of PSG+MRE in particular mental diseases.
Discussion
We identified RNA–miRNA competition in the human brain for duplicated PSG+MRE (0.48% of the total annotated PSGs) via their shared MREs with brain-expressed PCGs, not necessarily their parent genes. We also found a genomic selective sweep around PSG+MRE in the YRI population. RNA-seq analysis based on two independent human cohorts and two distinct RNA sequencing technologies, demonstrated higher expression in human brain neurons of PSG+MRE than PSG−MRE and apparent regulatory effects of PSG+MRE in the adult temporal lobe. These observations were supported by experimental manipulation tests and genomic analyses, were sustained after extensive tests of robustness and reflected stronger impact for PSG+MRE versus duplicated PSG−MRE compared to expressed versus non-expressed PSG−MRE.
PSG+MRE tend to be duplicated, and likely have functional promoter elements similar to those of their parent genes. Also, they show higher brain expression compared to processed PSG−MRE. This is compatible with the concept of expansion by gene duplication events of cognitive complexity;45 indicating that duplication events that led to non-coding genes could participate in human brain evolution. Also, human PSG+MRE tend to harbor less or different MREs than those of their parent genes, and show uncorrelated expression with their parent genes, as reported by others.46 However, our exemplary interference test indicated PSG+MRE-mediated miRNA destruction, which refers to a small subset of genes yet may be more powerful than sponging activity, and should be further studied for more PSG+MRE. Specifically, PSG+MRE and protein-coding genes with MRE overlaps (mostly non-PSG-parent gene pairs) presented highly correlated expression in the temporal lobe, supporting functional correlations between PSG+MRE with PCGs sharing MREs with them, but leaving the nature of these correlations open. Due to the human specificity of the observed phenomena, future in vivo tests might require ‘humanized’ mouse models; and expanded tests in larger sample cohorts can potentially strengthen the value of these observations.
Our global analysis spans in-house data sets and compiled data sets from very different biological origins, supporting our conclusion of a human brain-specific phenomenon. That PSG+MRE are more efficiently expressed in the brain than duplicated PSG−MRE may reflect both their young evolutionary age and their functional relevance. For example, impaired ACHE regulation by the primate-specific miRNA-608 causes massive whole-genome differences in numerous other brain targets and brain regions of miRNA-608,21 including the stress-related amygdala. Salmena et al.8 suggested that PSGs, or long non-coding RNAs, would be the main source for competitive miRNA targets to the ones located on their parent genes, which differs from our findings; and that PSGs would perform competition events to a higher extent than PCGs, which is supported by our findings. We further found that SNPs in PSG+MRE-adjacent regions associate with mental diseases, including autism, bipolar disorder or schizophrenia, and with differential expression of brain eQTLs. This is compatible with the genetic risk for mental diseases residing with common variation, with groups of SNPs of individually small effects having a substantial impact,47 and with expression change of a noncoding RNA in autistic patients contributing to the disease risk.19 However, these findings as well await further analysis and validation tests.
The detailed mechanisms by which each of the PSG+MRE contributes to the mental processes that are impaired in these diseases remain to be discovered, possibly by searching for de novo mutations in PSG+MRE genes in specific patient cohorts. For example, the HLA-C gene underwent recent human evolution, and we find it to interact with the highly expressed unrelated PSG-565 with which it shares multiple MREs. Also, ‘sponging’ and/or target-mediated miRNA destruction effects of miRNAs by long-non-coding RNAs32 and circular RNAs,48, 49 should be pursued as soon as full genomic coordinates for these RNAs will be established, especially in the cholinergic neurotransmission pathway50 which is relevant to many of the mental diseases. Further, recent reports indicate that some PSGs are translated into peptides in humans,51, 52 postulating an additional putative role for PSG+MRE. It will be interesting to find if a given PSG functions as a competitor and/or target-mediated destruction regulator in certain conditions, while producing peptides in others.
In conclusion, our study provides experimentally validated supportive evidence to the recent evolution of PSG+MRE and their current co-regulation of MRE-sharing coding transcripts in human brain neurons. This calls for further studies and suggests that balanced RNA competition and/or target-mediated miRNA destruction may contribute to human cognition and mental health, with new avenues for discovery of mental disease mechanisms, and for identifying novel genomic biomarkers and therapeutic targets for brain disorders.
Code availability
Upon request from barbashshahar@gmail.com
References
Boudreau RL, Jiang P, Gilmore BL, Spengler RM, Tirabassi R, Nelson JA et al. Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron 2014; 81: 294–305.
Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K et al. Birth and expression evolution of mammalian microRNA genes. Genome Res 2013; 23: 34–45.
Babbitt CC, Fedrigo O, Pfefferle AD, Boyle AP, Horvath JE, Furey TS et al. Both noncoding and protein-coding RNAs contribute to gene expression evolution in the primate brain. Genome Biol Evol 2010; 2: 67–79.
Oldham MC, Horvath S, Geschwind DH . Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci USA 2006; 103: 17973–17978.
Hu HY, He L, Fominykh K, Yan Z, Guo S, Zhang X et al. Evolution of the human-specific microRNA miR-941. Nat Commun 2012; 3: 1145.
Barbash S, Shifman S, Soreq H . Global coevolution of human MicroRNAs and their target genes. Mol Biol Evol 2014; 31: 1237–1247.
de la Mata M, Gaidatzis D, Vitanescu M, Stadler MB, Wentzel C, Scheiffele P et al. Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep 2015; 16: 500–511.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP . A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 2011; 146: 353–358.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP . A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010; 465: 1033–1038.
Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M . Assessing the ceRNA Hypothesis with Quantitative Measurements of miRNA and Target Abundance. Mol Cell 2014; 54: 766–776.
Haas G, Cetin S, Messmer M, Chane-Woon-Ming B, Terenzi O, Chicher J et al. Identification of factors involved in target RNA-directed microRNA degradation. Nucleic Acids Res 2016; 44: 2873–2887.
Nitzan M, Steiman-Shimony A, Altuvia Y, Biham O, Margalit H . Interactions between distant ceRNAs in regulatory networks. Biophys J 2014; 106: 2254–2266.
Lee DY, Jeyapalan Z, Fang L, Yang J, Zhang Y, Yee AY et al. Expression of versican 3'-untranslated region modulates endogenous microRNA functions. PLoS ONE 2010; 5: e13599.
Jeyapalan Z, Deng Z, Shatseva T, Fang L, He C, Yang BB . Expression of CD44 3'-untranslated region regulates endogenous microRNA functions in tumorigenesis and angiogenesis. Nucleic Acids Res 2011; 39: 3026–3041.
Bosson AD, Zamudio JR, Sharp PA . Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell 2014; 56: 347–359.
Jens M, Rajewsky N . Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet 2014; 16: 113–126.
Rippey C, Walsh T, Gulsuner S, Brodsky M, Nord AS, Gasperini M et al. Formation of chimeric genes by copy-number variation as a mutational mechanism in schizophrenia. Am J Hum Genet 2013; 93: 697–710.
Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science (New York, NY) 2008; 320: 539–543.
Kerin T, Ramanathan A, Rivas K, Grepo N, Coetzee GA, Campbell DB . A noncoding RNA antisense to moesin at 5p14.1 in autism. Sci Transl Med 2012; 4: 128ra140.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T et al. Strong association of de novo copy number mutations with autism. Science (New York, NY) 2007; 316: 445–449.
Hanin G, Shenhar-Tsarfaty S, Yayon N, Yau YH, Bennett ER, Sklan EH et al. Competing targets of microRNA-608 affect anxiety and hypertension. Hum Mol Genet 2014; 23: 4569–4580.
Lin T, Simchovitz A, Shenhar-Tsarfaty S, Vaisvaser S, Admon R, Hanin G et al. Intensified vmPFC surveillance over PTSS under perturbed microRNA-608/AChE interaction. Transl Psychiatry 2016; 6: e801.
Bennett DA, Schneider JA, Arvanitakis N, Wilson RS . Overview and findings from the religious orders study. Curr Alzheimer Res 2012; 9: 628–645.
Bennett DA, Schneider JA, Aggarwal NT, Arvanitakis Z, Shah RC, Kelly JF et al. Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 2006; 27: 169–176.
Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT et al. Natural history of mild cognitive impairment in older persons. Neurology 2002; 59: 198–205.
Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS . Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology 2005; 64: 834–841.
Gaidatzis D, van Nimwegen E, Hausser J, Zavolan M . Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinformatics 2007; 8: 69.
Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP . MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 2007; 27: 91–105.
Altshuler LL, Bartzokis G, Grieder T, Curran J, Jimenez T, Leight K et al. An MRI study of temporal lobe structures in men with bipolar disorder or schizophrenia. Biol Psychiatry 2000; 48: 147–162.
Salmond CH, Ashburner J, Connelly A, Friston KJ, Gadian DG, Vargha-Khadem F . The role of the medial temporal lobe in autistic spectrum disorders. Eur J Neurosci 2005; 22: 764–772.
Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR . Pseudogenes: pseudo-functional or key regulators in health and disease? RNA (New York, NY) 2011; 17: 792–798.
Soreq L, Guffanti A, Salomonis N, Simchovitz A, Israel Z, Bergman H et al. Long non-coding RNA and alternative splicing modulations in Parkinson's leukocytes identified by RNA sequencing. PLoS Comput Biol 2014; 10: e1003517.
Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci USA 2015; 112: 7285–7290.
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129: 823–837.
Guo JU, Agarwal V, Guo H, Bartel DP . Expanded identification and characterization of mammalian circular RNAs. Genome Biol 2014; 15: 409.
Cross-Disorder-Group-of-the-Psychiatric-Genomics-Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013; 381: 1371–1379.
Psychiatric-GWAS-Consortium-Bipolar-Disorder-Working-Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 2011; 43: 977–983.
Schizophrenia-Psychiatric-Genome-Wide-Association-Study-(GWAS)-Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet 2011; 43: 969–976.
Neale BM, Medland SE, Ripke S, Asherson P, Franke B, Lesch KP et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010; 49: 884–897.
Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013; 18: 497–511.
Sullivan PF, Daly MJ, O'Donovan M . Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet 2012; 13: 537–551.
Ben-David E, Shifman S . Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PLoS Genet 2012; 8: e1002556.
Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Irish-Schizophrenia-Genomics-Consortium-and-the-Wellcome-Trust-Case-Control-Consortium-2. Genome-wide association study implicates HLA-C*01:02 as a risk factor at the major histocompatibility complex locus in schizophrenia. Biol Psychiatry 2012; 72: 620–628.
Belgard TG, Geschwind DH . Retooling spare parts: gene duplication and cognition. Nat Neurosci 2013; 16: 6–8.
Kalyana-Sundaram S, Kumar-Sinha C, Shankar S, Robinson DR, Wu YM, Cao X et al. Expressed pseudogenes in the transcriptional landscape of human cancers. Cell 2012; 149: 1622–1634.
Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013; 45: 984–994.
Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014; 56: 55–66.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK et al. Natural RNA circles function as efficient microRNA sponges. Nature 2013; 495: 384–388.
Soreq H . Checks and balances on cholinergic signaling in brain and body function. Trends Neurosci 2015; 38: 448–458.
Ji Z, Song R, Regev A, Struhl K . Many lncRNAs, 5'UTRs, and pseudogenes are translated and some are likely to express functional proteins. eLife 2015; 4: e08890.
Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R et al. A draft map of the human proteome. Nature 2014; 509: 575–581.
Acknowledgements
We are grateful to Drs. Lilach Soreq, London and Eran Meshorer and David S. Greenberg, Jerusalem, for materials and fruitful discussions; to Dr Alexander Seitz and Dr Torsten Reda, Lexogen, Vienna, Mr. Alessandro Guffanti, Genomnia, Milan and Ms Geula Hanin, Ms Bettina Nadorp and Ms Michal Roitman, Jerusalem, for technical and analytic support; and to the Netherland Brain Bank and the Rush Alzheimer’s Disease Center, Chicago for tissues and data. This work was supported by the European Research Council Advanced Award (Grant number 321501, to HS), the Legacy Heritage Science Initiative (LHSI) of the Israel Science Foundation (Grant Number 378/11, to HS), The Austrian Research Promotion Agency (FFG Bridge1 project) (Grant number 853294 to HS) and by National Institutes of Health (Grant numbers P30AG10161, RF1AG15819, R01AG36042, R01AG36826 to DAB). SB was an incumbent of the TEVA National Network of Excellence in Neuroscience—NNE fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Barbash, S., Simchovitz, A., Buchman, A. et al. Neuronal-expressed microRNA-targeted pseudogenes compete with coding genes in the human brain. Transl Psychiatry 7, e1199 (2017). https://doi.org/10.1038/tp.2017.163
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2017.163
This article is cited by
-
Evolution of genetic networks for human creativity
Molecular Psychiatry (2022)
-
MicroRNA-298 reduces levels of human amyloid-β precursor protein (APP), β-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties
Molecular Psychiatry (2021)
-
A comparison between whole transcript and 3’ RNA sequencing methods using Kapa and Lexogen library preparation methods
BMC Genomics (2019)
-
The complex genetics and biology of human temperament: a review of traditional concepts in relation to new molecular findings
Translational Psychiatry (2019)
-
Effect of Sevoflurane Anesthesia on Brain Is Mediated by lncRNA HOTAIR
Journal of Molecular Neuroscience (2018)
