
Abstract
Studies of rare genetic variation have identified molecular pathways conferring risk for developmental neuropsychiatric disorders. To date, no published whole-exome sequencing studies have been reported in obsessive-compulsive disorder (OCD). We sequenced all the genome coding regions in 20 sporadic OCD cases and their unaffected parents to identify rare de novo (DN) single-nucleotide variants (SNVs). The primary aim of this pilot study was to determine whether DN variation contributes to OCD risk. To this aim, we evaluated whether there is an elevated rate of DN mutations in OCD, which would justify this approach toward gene discovery in larger studies of the disorder. Furthermore, to explore functional molecular correlations among genes with nonsynonymous DN SNVs in OCD probands, a protein–protein interaction (PPI) network was generated based on databases of direct molecular interactions. We applied Degree-Aware Disease Gene Prioritization (DADA) to rank the PPI network genes based on their relatedness to a set of OCD candidate genes from two OCD genome-wide association studies (Stewart et al., 2013; Mattheisen et al., 2014). In addition, we performed a pathway analysis with genes from the PPI network. The rate of DN SNVs in OCD was 2.51 × 10−8 per base per generation, significantly higher than a previous estimated rate in unaffected subjects using the same sequencing platform and analytic pipeline. Several genes harboring DN SNVs in OCD were highly interconnected in the PPI network and ranked high in the DADA analysis. Nearly all the DN SNVs in this study are in genes expressed in the human brain, and a pathway analysis revealed enrichment in immunological and central nervous system functioning and development. The results of this pilot study indicate that further investigation of DN variation in larger OCD cohorts is warranted to identify specific risk genes and to confirm our preliminary finding with regard to PPI network enrichment for particular biological pathways and functions.
Similar content being viewed by others

Introduction
The etiology of common neuropsychiatric disorders is believed to be multifactorial, with contributions from both environmental and genetic factors.1 Studies have predominantly focused on common polymorphic variants, finding small effect sizes, and replication of positive findings has been difficult.2 This raises the question of where the ‘missing heritability’ of complex diseases might be found. A substantial portion may indeed reflect gene–gene interactions and gene–environmental interactions that have not been taken into consideration in estimates of narrow sense heritability.3 In addition, understanding the heritability of genetic diseases requires a more comprehensive assessment of human genetic variation, including rare variation, throughout the genome.3, 4
The emergence of next-generation sequencing platforms is facilitating comprehensive searches for both rare and common single-nucleotide variants (SNVs) across all genes in the genome via whole-exome sequencing (WES).5 Although protein-coding genes constitute only about 1% of the human genome, they are estimated to harbor 85% of mutations with large effects in disease-related traits.6
Several groups have identified genes conferring risk for intellectual disability,7, 8 schizophrenia9, 10 and autism11, 12, 13, 14 by applying WES in families with no previous history of these disorders or their related phenotypes—so-called sporadic or simplex families—and identifying recurrent de novo (DN) SNVs. The DN SNVs tend to be more common in patients than in controls or unaffected siblings, mainly when such variations are nonsynonymous and located in brain-expressed genes.10, 11, 15
Obsessive-compulsive disorder (OCD) is a severe neuropsychiatric disorder, commonly having an early age of onset, and is characterized by the presence of obsessions (unwanted, intrusive thoughts) and compulsions (repetitive behaviors) that can become incapacitating.16, 17 Family, twin, segregation and linkage studies suggest a complex genetic etiology, complicating the confirmation of specific risk variants; consequently, the cellular and molecular mechanisms underlying OCD pathophysiology remain uncertain.18
A meta-analysis of common variant genetic association studies of OCD found multiple polymorphisms with significant association.19 Significant variants were located in serotonergic genes, and, in males only, catecholamine modulation genes.19 The first published genome-wide association study (GWAS), querying common polymorphisms in a large cohort of OCD patients, did not find any variants reaching the genome-wide significance threshold, but top signals in several common variants were related to transcriptional regulation, cytoskeleton dynamics, ion channel assembly and gating, protein ubiquitination and degradation, and glutamate signaling.20 The latest GWAS in OCD21 also found no variants reaching genome-wide significance but showed significant overlap with top signals from the first GWAS. Furthermore, network analysis of top signals in these GWAS studies support the idea that genetic risk for OCD may cluster in certain biological networks or systems.22
There have been few studies examining rare variants in OCD.23, 24, 25, 26, 27, 28, 29, 30, 31 The first genome-wide investigation of rare copy number variation (CNV) in OCD and Tourette syndrome reported a 3.3-fold increase in large (>500 kb) deletions that overlap with CNVs reported in other neurodevelopmental disorders. An overall DN CNV rate of 1.4% was reported in OCD, slightly higher than the estimated frequency for controls, suggesting that rare DN variation may have a role in OCD pathogenesis.32 To date, there have been no published studies examining rare coding SNVs across the genome in OCD.
In the current pilot study, we examined 20 simplex OCD parent–child trios using WES to detect SNVs. Given the recent success in other neuropsychiatric disorders, we focused on detection of DN SNVs in OCD, aiming to advance our understanding of the contribution of rare nonsynonymous DN SNVs to the disorder. Next, we mapped these SNVs onto a protein–protein interaction (PPI) network, hypothesizing that perturbations of integrated molecular networks through genomic and environmental influences can increase the risk for complex diseases such as OCD33 and that topological properties of PPI networks represent molecular functional correlations among genes that are important for understanding disease biology. We then performed Degree-Aware Disease Gene Prioritization (DADA), ranking our SNVs against ‘seed’ genes identified from two OCD GWAS, predicting that top-ranked genes in this analysis would achieve greater connectivity in our PPI network. Finally, we asked whether all the genes in our PPI network were enriched in certain canonical biological pathways, in an attempt to enhance our understanding of OCD pathophysiology.
Materials and methods
Subjects
This study was approved by the Research Ethics Committees of the University of São Paulo School of Medicine, as well as by the Brazilian National Commission of Research Ethics (CONEP, process number: 16756). All the participating subjects gave written informed consent.
The OCD patients, meeting DSM-IV criteria for the diagnosis, and their unaffected parents, were recruited at the Outpatient Clinic of the Obsessive-Compulsive Spectrum Disorders Program of the Institute of Psychiatry, at the University of São Paulo School of Medicine Hospital das Clínicas. The probands were evaluated by semi-structured and structured interviews included in the Brazilian Research Consortium on Obsessive-Compulsive Spectrum Disorders instruments, administered by trained clinicians (Supplementary Methods).34 The parents were directly screened with the Structured Clinical Interview for DSM-IV Axis I Disorders; those with any Axis I psychiatric diagnosis were excluded.
Capture and sequencing
Exome capture, sequencing and variant detection were performed at the Yale Center for Genomic Analysis, as described previously11 and summarized below.
The DNA samples from whole blood were enriched for exonic sequence with the NimbleGen SeqCap EZ Exome v2 capture library (Roche NimbleGen, Madison, WI, USA). The samples were sequenced using the Illumina HiSeq 2000 platform (74 bp paired-end reads; Illumina, San Diego, CA, USA). We multiplexed four samples during each capture reaction and sequencing lane, pooling parents and probands when possible.
Sequence alignment and variant calling
Short-read sequences were aligned to the human reference genome (hg19/NCBI 37) using the Burrows–Wheeler Aligner (http://bio-bwa.sourceforge.net/).35 The aligned reads were trimmed to the exome target using an in-house script. The trimmed and aligned data were converted to a sorted binary format, and duplicates were removed using SAMtools (http://samtools.sourceforge.net/),36 which was also used to identify the SNVs. SAMtools was selected for initial variant calling in order to replicate the bioinformatic pipeline used to analyze control subjects in the literature, which served as our comparison cohort for the overall rate of DN SNVs.11 For subsequent analyses of PPI networks, pathways and disease gene prioritization, we also considered confirmed variants from a second alignment and variant calling pipeline which followed the GATK v3 best practices guidelines (Supplementary Methods). The purpose of adding variants from this second pipeline was to ensure discovery of the maximum number of DN variants for our downstream analyses. The variants from both pipelines were annotated against the UCSC gene definitions (http://genome.ucsc.edu/) for impact on the encoded protein (silent, missense, nonsense and splice site) and for population allele frequency using ExAC v0.3.37
Family relatedness check
A panel of informative genotypes was used to perform identity-by-descent in each study subject using the PLINK software package (http://pngu.mgh.harvard.edu/~purcell/plink/).38 Families were discarded if expected relationships did not confirm.
DN SNVs
SNVs were predicted to be DN if all the following criteria were met: (1) the variant was not predicted in either parent, (2) at least eight unique reads supported the variant in the proband and (3) the locus had ⩾20 × coverage in both parents and the proband. We validated all the DN SNVs by PCR and Sanger sequencing (Supplementary Methods). We calculated the per-base rate of DN SNVs in our OCD samples and compared with rates reported in several published studies of reference populations and psychiatric disorders using a Poisson test (Supplementary Methods).
PPI network analysis
Next, we mapped interactions between genes harboring validated nonsynonymous (missense or nonsense) DN SNVs by constructing a PPI network. A network is a set of elements interacting with each other through pairwise interactions. In the case of biological PPI networks, the components are the gene proteins (nodes) that are connected to each other by links (edges) representing known physical interactions between two components. We used Cytoscape39 and the iRefScape plugin40 to create a PPI network. This network is based on databases of direct physical interactions between genes, with proteins as nodes and interactions as undirected edges.41 We included PPIs that were present in any of the 10 databases consolidated within iRefIndex40 (Supplementary Methods). We applied two methods of permutation, ‘seed randomization’ and ‘network permutation,’ using GeneNet toolbox for MATLAB42 to show that the connectivity metrics of our network are different from networks generated by chance.
DADA analysis
To investigate the potential relevance of our DN SNVs in OCD, we applied Degree-Aware Disease Gene Prioritization (DADA; http://compbio.case.edu/dada/), a tool for performing network-based prioritization of candidate disease genes.43 DADA analysis of our candidate genes harboring validated DN SNVs detected by WES were performed with the ‘seed’ genes (curated from other studies as likely to be associated with the disorder) from two OCD GWAS reported to date20, 21 (Supplementary Methods). Prioritization is made based on the proximity of interaction (protein–protein interaction) of these seed genes with our WES candidate genes, using the NCBI Entrez Gene Database, which integrates human protein–protein interaction network data from several other databases such as HPRD, BioGRID and BIND.44
Pathway analysis
To determine whether the list of all genes identified in our PPI network showed enrichment for specific biological pathways, we used Ingenuity Pathway Analysis (build version 355958M, content version 24718999; Ingenuity Systems, http://www.ingenuity.com/) to map genes to canonical pathways (Supplementary Methods). We also examined whether our confirmed DN missense variants in OCD were enriched for published lists of genes believed to contribute risk for autism, schizophrenia and intellectual disability45 using DNENRICH.46 Finally, we asked whether our PPI network gene lists showed enrichment for these disorders, calculating P-values in R (function phyper) based on a hypergeometric distribution.
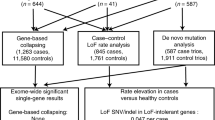
A summary of all the methods and analyses is shown in Figure 1.

Single-nucleotide variant (SNV) discovery, quality control, annotation and analysis workflow. Whole-blood samples from obsessive-compulsive disorder (OCD) probands and their unaffected parents were enriched for exonic sequence with the NimbleGen SeqCap EZ Exome capture reagents and sequenced using the Illumina HiSeq 2000 platform. Identity by descent analysis was performed to confirm relatedness among samples. Final analyses included 17 OCD trios. Only de novo (DN) SNVs called by SAMtools and validated by Sanger sequencing (present in proband and absent in parents) were carried into DN SNV rate analyses. For subsequent analyses of protein–protein interaction (PPI), Degree-Aware Disease Gene Prioritization (DADA) and Ingenuity Pathway Analyses (IPA), we also included confirmed DN SNVs from a second alignment and variant calling pipeline, which followed the GATK v3 best practices guidelines.
Results
Subjects
Our final analysis included WES data from 17 parent–child trios, each trio consisting of a child with OCD and their unaffected parents. Although we started with 20 trios, three were omitted from the analysis due to failing the family relatedness check. Among the 17 OCD trios included in our analyses, the mean age at symptom onset was 8.6 (±2.7) years and the mean Y-BOCS score was 26.1 (±5.7). Other clinical and demographic characteristics of included subjects are shown in Supplementary Table 1.
Sequencing
On average, 91.6% of the generated sequence aligned to the reference genome, and 96.7% of the targeted bases in each individual were assessed by ⩾8 independent sequence reads. Only those bases with ⩾20 × coverage in all family members were considered for DN SNV detection, allowing for analysis of de novo events in 92.8% of all targeted bases (Supplementary Table 2).
DN SNVs
Using the SAMtools variant detection pipeline (for comparison of DN SNV rate in OCD versus controls reported in the literature), 19 DN SNVs (12 nonsynonymous, 7 silent) were confirmed by Sanger sequencing (Table 1, Figure 1). Among nonsynonymous DN SNVs, we confirmed one nonsense mutation and 11 missense mutations. Of the 17 patients, 8 (~47%) carried at least one nonsynonymous DN SNV.
The total number of coding base pairs screened at ⩾20 × was 756.8 Mbp (Supplementary Table 3). Nineteen DN SNVs were validated, corresponding to a rate of 2.51 × 10−8 per base pair for human haploid genome and 1.12 events per trio. A two-tailed Poisson rate ratio test (R package rateratio.test) indicated that the rate of DN SNVs observed in our study differs significantly from that observed in unaffected siblings of autism probands, sequenced on the same platform and analyzed with the same bioinformatics pipeline11 (P=0.02, Table 2). There was no significant difference in paternal ages at conception between our OCD (mean 30.2 years) and this control cohort (mean 32.2 years; P=0.22, two-tailed Mann–Whitney test; Supplementary Table 4). We did not observe a significant difference when comparing our OCD DN SNV rate with those previously reported in schizophrenia,10, 46 autism11, 13, 47, 48 and intellectual disability7, 8 studies (Table 2).
Using the GATK v3 best practices variant calling pipeline, we confirmed eight additional nonsynonymous (all missense) DN SNVs, which we included in downstream analyses, for a total of 20 nonsynonymous DN SNVs (Table 1, Figure 1). There were no genes that harbored more than one confirmed DN SNV among the OCD subjects.
PPI network and DADA analyses
To construct the PPI network, we selected the genes with nonsynonymous (missense, nonsense) DN SNVs detected by our GATK and SAMtools pipelines (Figure 1, Table 1). Of the 20 genes harboring confirmed nonsynonymous DN SNVs, six genes (FAM5B, CCDC108, VCX2, MUC5B, ARHGAP6, SLC35G5) were not present in the PPI databases. Therefore, 14 genes served as the input for construction of our PPI network (Table 1). In the resulting PPI network of 320 nodes, two genes from our original inputs (WWP1, SMAD4) were found to be highly and independently interconnected with other non-neighboring genes (that is, they were found to be ‘brokers’), displaying 40 and 187 interactions (edges), respectively; six genes were found to be ‘bottleneck’ genes (WWP1, SMAD4, CR1, AP1G1, MYO10, SNUPN) that connect different complexes or pathways in the network; and three were found to be ‘bridge’ genes (BAMBI, ABCE1, NDE1) with high information flow, located between highly connected modules. (Figure 2, Supplementary Figure 1, Supplementary Table 5). Network permutations showed that connectivity metrics of ‘seed indirect degrees’ mean and ‘common connectors means’ for our network are different from these metrics in the permuted networks (1000 permutations).

Protein–protein interaction network including nonsynonymous DN SNVs in OCD. Genes connecting components of the protein–protein interaction network harbor DN SNVs among the obsessive-compulsive disorder probands (red circles). Genes shaded blue are bridges, linking well-connected regions of the PPI network. Genes shaded orange are brokers, having a large number of connections with non-neighboring genes. Genes within red squares are bottlenecks, connecting different parts of the network with high betweenness. DN SNV, de novo single-nucleotide variant; OCD, obsessive-compulsive disorder; PPI, protein–protein interaction.
Next, we performed DADA analysis to rank our 14 nonsynonymous DN SNVs in OCD subjects, using top genes from two OCD GWAS20, 21 as seed genes (Supplementary Table 6) to rank our DN SNVs. Genes with the highest prioritization rank using seeds from the first GWAS20, 21 were BAMBI, followed by SMAD4 and WWP1. Using seed genes from the second GWAS,20, 21 highest prioritization rank was seen for SMAD4, followed by WWP1 and MYO10. Of these four genes ranked highly by DADA, two are considered broker genes (WWP1, SMAD4) in our PPI network, three are considered bottleneck genes (WWP1, SMAD4, BAMBI, MYO10), and one is considered a bridge gene (BAMBI; Supplementary Table 5).
Pathway analyses
Finally, a pathway analysis was performed to determine whether the nodes in our PPI network are enriched in canonical pathways from the Ingenuity Pathway Analysis database. We performed two pathway analyses using different gene lists as input: (1) using all 320 nodes in the PPI network (Supplementary Table 5) and (2) using 37 nodes from the PPI network determined to be brokers, bridges or bottlenecks by measurements of topological centrality (Supplementary Table 5), as these nodes may carry greater functional significance.49, 50 The analysis of all PPI network nodes found significant enrichment for pathways related to transforming growth factor beta (TGF-β) signaling, bone morphogenic protein signaling and glucocorticoid receptor signaling (Table 3). Narrowing the input list to bridges, brokers and bottlenecks also yields enrichment in TGF-β and glucocorticoid receptor signaling. Functional network enrichment for these central PPI nodes includes embryonic development, cell-to-cell signaling, cell death and survival, and cellular function and maintenance (Supplementary Table 9). There was no significant overlap between these same gene lists and lists of risk genes for autism, schizophrenia and intellectual disability45 (Supplementary Table 10).
Discussion
To our knowledge, this is the first reported WES study in OCD designed to search for rare DN SNVs across all the coding regions of the genome in parent–child trios. As with similar studies in other neuropsychiatric disorders, the DN SNVs we identified may include true OCD risk variants and point toward relevant gene networks and canonical pathways. The small number of subjects in the present study precludes confirmation of the involvement of specific genes or variants in OCD. Nevertheless, the finding that DN SNVs occur more frequently in OCD is an essential prelude to identifying specific risk genes through the identification of recurrent mutations (independent DN variants mapping within the identical gene or at the same chromosomal locus) in larger patient cohorts. Furthermore, the network analyses in this pilot study integrate prior findings from GWAS20, 21 to generate hypotheses of potentially relevant genes, biological pathways, networks and processes in OCD that can be tested in larger studies.
Our first analysis used a SAMtools variant detection pipeline to compare the rate of confirmed DN SNVs in our OCD samples versus published rates in controls and other disorders. Our observed per base pair per generation DN SNV rate (2.51 × 10−8) differs significantly from a rate previously reported in unaffected subjects (1.31 × 10−8; ref. 11) using an identical variant calling pipeline. Parental age does not seem to account for this difference, and we did not find any difference between our rate and those reported in schizophrenia,10, 46 autism11, 13, 47, 48 or intellectual disability7, 8 exome-sequencing studies (Table 2).
In addition to the variants conformed using SAMtools, we confirmed eight additional nonsynonymous DN SNVs, predicted using a GATK best practices pipeline. We used all 20 nonsynonymous DN SNVs detected by both pipelines for downstream analyses. Given that protein gene products associated with disease have a higher likelihood of physically interacting,41, 51, 52 we next constructed a PPI network starting with candidate genes harboring confirmed nonsynonymous DN SNVs in OCD. Two of our candidate genes were classified as ‘brokers’ (that is, highly and independently connected with non-neighboring genes) in this analysis, suggesting that they may be more relevant for disease (Supplementary Table 5).52 WWP1 (WW domain containing E3 ubiquitin protein ligase 1) inhibits transcriptional activity induced by TGF-β, a member of a highly pleiotropic cytokine family that maintains immune homeostasis, directs lymphocyte differentiation and orchestrates aspects of embryonic development including neuronal migration and synapse formation.53, 54 SMAD4 (SMAD family member 4) codes for a signal transduction protein that is activated by TGF-β signaling during proliferation and differentiation of the central nervous system.55 Furthermore, some of our candidate genes were classified as ‘bottlenecks’ (key connectors) in the PPI network (Supplementary Table 5), believed to be one of the most significant indicators of essentiality.56 Aside from WWP1 and SMAD4, other bottleneck genes were CR1 (complement component (3b/4b) receptor 1 [Knops blood group]), an important member of the family of regulators of complement activation and a crucial multifunctional mediator of innate immunity; MYO10 ( myosin X), which may have a role in neurite outgrowth and axon guidance;57 and AP1G1 (adaptor-related protein complex 1, gamma 1 subunit), important for the formation of clathrin-coated vesicles to transport ligand–receptor complexes from the plasma membrane or from the trans-Golgi network to lysosomes.58 Although AP1G1 has not been associated with any psychiatric disorder, it is noteworthy that the first genome-wide investigation of large, rare CNVs in OCD found a de novo CNV encompassing AP1GBP1, a related gene that also acts on clathrin-coated vesicles and may have a role in endocytosis.32
To investigate the potential relevance of our PPI network to OCD, we conducted a DADA analysis to see whether prioritized genes would have more connectivity in our PPI network. In this analysis, using a seed gene list curated from the two GWAS in OCD,20, 21 two of the three highest ranked genes (WWP1 and SMAD4) were found to be brokers in the PPI network (Supplementary Table 7). The highest ranked gene in the DADA analysis using GWAS I (Supplementary Table 7) was BAMBI (bone morphogenetic protein and activin membrane-bound inhibitor), coding for a pseudoreceptor that negatively modulates TGF-β.59 BAMBI is classified as a bridge gene in our PPI network and is among the genes regulated by top-ranking SNPs in the OCD GWAS I secondary analysis;20 its protein product has been found to be selectively and significantly enriched in white matter progenitor cells, and can modulate their differentiation in oligodendrocytes or astrocytes.60
Furthermore, it is notable that the top-ranked genes in the DADA analyses (BAMBI, SMAD4, WWP1, MYO10, AP1G1, ATP2B2) have negative Residual Variation Intolerance Scores,61 indicating that in large exome-sequencing databases, they are found to contain less common functional genetic variation relative to the genome-wide expectation. Genes with negative Residual Variation Intolerance Scores are referred to as relatively ‘intolerant’ to genetic variation and more likely to be involved with neurodevelopmental disease61 (Table 1, Supplementary Tables 7 and 8).
The canonical pathway analysis results from all the 320 nodes in our PPI network show enrichment for TGF-β signaling, bone morphogenic protein signaling and glucocorticoid signaling (Table 3); narrowing this list to 37 nodes classified as bridges, brokers and bottlenecks (Supplementary Table 5) shows enrichment for TGF-β and glucocorticoid receptor signaling. TGF-β signaling is mediated by a family of structurally related cytokines and by SMAD proteins that act to control proliferation, differentiation, migration and apoptosis of many different cell types. Bone morphogenic proteins are members of the TGF-β protein family of extracellular ligands,62 important in the neuronal protection against both apoptosis and excitotoxicity.63 The fidelity of these pathways are crucial for normal nervous system development and their disruption has been suggested to underlie schizophrenia pathology.63 Finally, glucocorticoids, a major subclass of steroid hormones, regulate a large number of immune, metabolic, cardiovascular and behavioral functions. Their major effects are anti-inflammatory via transcription induction of anti-inflammatory genes and by repression of inflammatory genes. Furthermore, their anti-inflammatory actions are complemented by their ability to induce apopotosis of cells, including monocytes and T lymphocytes. Imbalances in this pathway have been suggested in OCD, anxiety and other neuropsychiatric disorders.64, 65, 66
The major limitation of the current study is the small sample size. When studying rare variants, larger samples are needed to adequately show that certain genes or variants are associated with the disorder. Therefore, our study should be considered preliminary, requiring replication in larger OCD cohorts. Recruitment for a larger investigation of DN exome sequence variation in OCD trios is currently underway.
Although we are unable to pinpoint definitive risk genes or variants in a study of this size, we were able to show, for the first time, that (1) sporadic OCD families may have higher rates of DN coding SNVs, suggesting that further study of this type of variation in larger cohorts holds potential to identify risk genes; (2) a PPI network can be constructed on the basis of DN SNVs that appears to have relevance to OCD, judged against common variant findings from other genetic studies of OCD; and (3) PPI network genes show enrichment for biological pathways and functions involved with neurodevelopmental and immunological processes. These findings hold great promise for elucidating our understanding of OCD neurobiology and potential treatments, and deserve further scrutiny in larger cohorts.
References
State MW, Levitt P . The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci 2011; 14: 1499–1506.
Visscher PM, Brown MA, McCarthy MI, Yang J . Five years of GWAS discovery. Am J Hum Genet 2012; 90: 7–24.
Zuk O, Hechter E, Sunyaev SR, Lander ES . The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci USA 2012; 109: 1193–1198.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ et al. Finding the missing heritability of complex diseases. Nature 2009; 461: 747–753.
Buxbaum JD, Daly MJ, Devlin B, Lehner T, Roeder K, State MW et al. The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron 2012; 76: 1052–1056.
Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA 2009; 106: 19096–19101.
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012; 367: 1921–1929.
Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012; 380: 1674–1682.
Xu B, Roos JL, Dexheimer P, Boone B, Plummer B, Levy S et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet 2011; 43: 864–868.
Girard SL, Gauthier J, Noreau A, Xiong L, Zhou S, Jouan L et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat Genet 2011; 43: 860–863.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012; 485: 237–241.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011; 43: 585–589.
O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012; 485: 246–250.
Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet 2012; 8: e1002635.
Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR et al. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet 2010; 87: 316–324.
Ayuso-Mateos JL . Global Burden of Obsessive-Compulsive Disorder in the Year 2000. World Health Organization: Geneva, http://www.who.int/healthinfo/statistics/bod_obsessivecompulsive.pdf, 2006.
Miguel EC, Leckman JF, Rauch S, do Rosario-Campos MC, Hounie AG, Mercadante MT et al. Obsessive-compulsive disorder phenotypes: implications for genetic studies. Mol Psychiatry 2005; 10: 258–275.
Pauls DL . The genetics of obsessive-compulsive disorder: a review. Dialogues Clin Neurosci 2010; 12: 149–163.
Taylor S . Molecular genetics of obsessive-compulsive disorder: a comprehensive meta-analysis of genetic association studies. Mol Psychiatry 2012; 18: 799–805.
Stewart SE, Yu D, Scharf JM, Neale BM, Fagerness JA, Mathews CA et al. Genome-wide association study of obsessive-compulsive disorder. Mol Psychiatry 2013; 18: 788–798.
Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT et al. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry 2014; 20: 337–344.
Pauls DL, Abramovitch A, Rauch SL, Geller DA . Obsessive-compulsive disorder: an integrative genetic and neurobiological perspective. Nat Rev Neurosci 2014; 15: 410–424.
Moya PR, Dodman NH, Timpano KR, Rubenstein LM, Rana Z, Fried RL et al. Rare missense neuronal cadherin gene (CDH2) variants in specific obsessive-compulsive disorder and Tourette disorder phenotypes. Eur J Hum Genet 2013; 21: 850–854.
Hooper SD, Johansson AC, Tellgren-Roth C, Stattin EL, Dahl N, Cavelier L et al. Genome-wide sequencing for the identification of rearrangements associated with Tourette syndrome and obsessive-compulsive disorder. BMC Med Genet 2012; 13: 123.
Veenstra-VanderWeele J, Xu T, Ruggiero AM, Anderson LR, Jones ST, Himle JA et al. Functional studies and rare variant screening of SLC1A1/EAAC1 in males with obsessive-compulsive disorder. Psychiatr Genet 2012; 22: 256–260.
Walitza S, Bove DS, Romanos M, Renner T, Held L, Simons M et al. Pilot study on HTR2A promoter polymorphism, -1438G/A (rs6311) and a nearby copy number variation showed association with onset and severity in early onset obsessive-compulsive disorder. J Neural Transm (Vienna) 2012; 119: 507–515.
Delorme R, Moreno-De-Luca D, Gennetier A, Maier W, Chaste P, Mössner R et al. Search for copy number variants in chromosomes 15q11-q13 and 22q11.2 in obsessive compulsive disorder. BMC Med Genet 2010; 11: 100.
Ozomaro U, Cai G, Kajiwara Y, Yoon S, Makarov V, Delorme R et al. Characterization of SLITRK1 variation in obsessive-compulsive disorder. PLoS One 2013; 8: e70376.
Han L, Nielsen DA, Rosenthal NE, Jefferson K, Kaye W, Murphy D et al. No coding variant of the tryptophan hydroxylase gene detected in seasonal affective disorder, obsessive-compulsive disorder, anorexia nervosa, and alcoholism. Biol Psychiatry 1999; 45: 615–619.
Wang Y, Adamczyk A, Shugart YY, Samuels JF, Grados MA, Greenberg BD et al. A screen of SLC1A1 for OCD-related alleles. Am J Med Genet B Neuropsychiatr Genet 2010; 153B: 675–679.
Cappi C, Hounie AG, Mariani DB, Diniz JB, Silva AR, Reis VN et al. An inherited small microdeletion at 15q13.3 in a patient with early- onset obsessive-compulsive disorder. PLoS One 2014; 9: e110198.
McGrath LM, Yu D, Marshall C, Davis LK, Thiruvahindrapuram B, Li B et al. Copy number variation in obsessive-compulsive disorder and tourette syndrome: a cross-disorder study. J Am Acad Child Adolesc Psychiatry 2014; 53: 910–919.
Lage K . Protein-protein interactions and genetic diseases: the interactome. Biochim Biophys Acta 2014; 1842: 1971–1980.
Miguel EC, Ferrão YA, Rosário MC, Mathis MA, Torres AR, Fontenelle LF et al. The Brazilian Research Consortium on Obsessive-Compulsive Spectrum Disorders: recruitment, assessment instruments, methods for the development of multicenter collaborative studies and preliminary results. Rev Bras Psiquiatr 2008; 30: 185–196.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T et al. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv 2015; doi:10.1101/123456.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, Lotia S et al. A travel guide to Cytoscape plugins. Nat Methods 2012; 9: 1069–1076.
Razick S, Mora A, Michalickova K, Boddie P, Donaldson IM, iRefScape. A . Cytoscape plug-in for visualization and data mining of protein interaction data from iRefIndex. BMC Bioinformatics 2011; 12: 388.
Barabási AL, Gulbahce N, Loscalzo J . Network medicine: a network-based approach to human disease. Nat Rev Genet 2011; 12: 56–68.
Taylor A, Steinberg J, Andrews TS, Webber C . GeneNet Toolbox for MATLAB: a flexible platform for the analysis of gene connectivity in biological networks. Bioinformatics 2015; 31: 442–444.
Erten S, Bebek G, Ewing RM, Koyutürk M, DADA . Degree-aware algorithms for network-based disease gene prioritization. BioData Min 2011; 4: 19.
Brown GR, Hem V, Katz KS, Ovetsky M, Wallin C, Ermolaeva O et al. Gene: a gene-centered information resource at NCBI. Nucleic Acids Res 2015; 43 (Database issue): D36–D42.
Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014; 515: 216–221.
Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014; 506: 179–184.
Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012; 485: 242–245.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012; 74: 285–299.
Joy MP, Brock A, Ingber DE, Huang S . High-betweenness proteins in the yeast protein interaction network. J Biomed Biotechnol 2005; 2005: 96–103.
Khuri S, Wuchty S . Essentiality and centrality in protein interaction networks revisited. BMC Bioinformatics 2015; 16: 1–8.
Barabási AL, Oltvai ZN . Network biology: understanding the cell's functional organization. Nat Rev Genet 2004; 5: 101–113.
Cai JJ, Borenstein E, Petrov DA . Broker genes in human disease. Genome Biol Evol 2010; 2: 815–825.
Heupel K, Sargsyan V, Plomp JJ, Rickmann M, Varoqueaux F, Zhang W et al. Loss of transforming growth factor-beta 2 leads to impairment of central synapse function. Neural Dev 2008; 3: 25.
Goines PE, Ashwood P . Cytokine dysregulation in autism spectrum disorders (ASD): possible role of the environment. Neurotoxicol Teratol 2013; 36: 67–81.
Falk S, Joosten E, Kaartinen V, Sommer L . Smad4 and Trim33/Tif1γ redundantly regulate neural stem cells in the developing cortex. Cereb Cortex 2014; 24: 2951–2963.
Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M . The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol 2007; 3: e59.
Lai M, Guo Y, Ma J, Yu H, Zhao D, Fan W et al. Myosin X regulates neuronal radial migration through interacting with N-cadherin. Front Cell Neurosci 2015; 9: 326.
Hartmann-Stühler C, Prange R . Hepatitis B virus large envelope protein interacts with gamma2-adaptin, a clathrin adaptor-related protein. J Virol 2001; 75: 5343–5351.
Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massagué J et al. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999; 401: 480–485.
Sim FJ, Lang JK, Waldau B, Roy NS, Schwartz TE, Pilcher WH et al. Complementary patterns of gene expression by human oligodendrocyte progenitors and their environment predict determinants of progenitor maintenance and differentiation. Ann Neurol 2006; 59: 763–779.
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB . Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 2013; 9: e1003709.
Liu A, Niswander LA . Bone morphogenetic protein signalling and vertebrate nervous system development. Nat Rev Neurosci 2005; 6: 945–954.
Jia P, Wang L, Meltzer HY, Zhao Z . Common variants conferring risk of schizophrenia: a pathway analysis of GWAS data. Schizophr Res 2010; 122: 38–42.
Grabe HJ, Freyberger HJ, Maier W . Obsessive-compulsive symptom exacerbation following cortisone treatment. Neuropsychobiology 1998; 37: 91–92.
Sulkowski ML, Geller DA, Lewin AB, Murphy TK, Mittelman A, Brown A et al. The future of D-cycloserine and other cognitive modifiers in obsessive-compulsive and related disorders. Curr Psychiatry Rev 2014; 10: 317–324.
Tajima-Pozo K, Montes-Montero A, Guemes I, Gonzalez-Vives S, Diaz-Marsa M, Carrasco JL . [Contributions of cortisol suppression tests to understanding of psychiatric disorders: a narrative review of literature]. Endocrinol Nutr 2013; 60: 396–403.
Acknowledgements
We thank Dr Marcelo Batistuzzo, Dr Daniel L Costa and Dr Michael Bloch for their helpful comments on the manuscript. We also thank the families and patients who participated in this research. This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, São Paulo Research Foundation—process: 2008/11537-7; and process: 2011/14658-2); the Brazilian Instituto Nacional de Psiquiatria do Desenvolvimento para Infância e Adolescência (INPD, National Institute of Developmental Psychiatry for Children and Adolescents).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Cappi, C., Brentani, H., Lima, L. et al. Whole-exome sequencing in obsessive-compulsive disorder identifies rare mutations in immunological and neurodevelopmental pathways. Transl Psychiatry 6, e764 (2016). https://doi.org/10.1038/tp.2016.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2016.30
This article is cited by
-
Gene expression study in monocytes: evidence of inflammatory dysregulation in early-onset obsessive-compulsive disorder
Translational Psychiatry (2022)
-
Immunological causes of obsessive-compulsive disorder: is it time for the concept of an “autoimmune OCD” subtype?
Translational Psychiatry (2022)
-
The phenotypic spectrum and genotype-phenotype correlations in 106 patients with variants in major autism gene CHD8
Translational Psychiatry (2022)
-
A dimensional perspective on the genetics of obsessive-compulsive disorder
Translational Psychiatry (2021)
-
Serotonin 5-HT1B receptor-mediated behavior and binding in mice with the overactive and dysregulated serotonin transporter Ala56 variant
Psychopharmacology (2021)

{kind=link}