
Abstract
Several gene expression experiments on autism spectrum disorders have been conducted using both blood and brain tissue. Individually, these studies have advanced our understanding of the molecular systems involved in the molecular pathology of autism and have formed the bases of ongoing work to build autism biomarkers. In this study, we conducted an integrated systems biology analysis of 9 independent gene expression experiments covering 657 autism, 9 mental retardation and developmental delay and 566 control samples to determine if a common signature exists and to test whether regulatory patterns in the brain relevant to autism can also be detected in blood. We constructed a matrix of differentially expressed genes from these experiments and used a Jaccard coefficient to create a gene-based phylogeny, validated by bootstrap. As expected, experiments and tissue types clustered together with high statistical confidence. However, we discovered a statistically significant subgrouping of 3 blood and 2 brain data sets from 3 different experiments rooted by a highly correlated regulatory pattern of 66 genes. This Root 66 appeared to be non-random and of potential etiologic relevance to autism, given their enriched roles in neurological processes key for normal brain growth and function, learning and memory, neurodegeneration, social behavior and cognition. Our results suggest that there is a detectable autism signature in the blood that may be a molecular echo of autism-related dysregulation in the brain.
Similar content being viewed by others


Introduction
Autism is regarded as one condition among a genetically heterogeneous group of neurodevelopmental syndromes with high prevalence1 that has a wide range of phenotypes, collectively grouped together as autism spectrum disorder (ASD). The unifying clinical features across the spectrum involve fundamental impairments in social interaction, communication deficits and highly restrictive interest and/or repetitive behaviors.2, 3 Although there is no unifying hypothesis about the molecular pathology of autism, it is clear that the disorder is highly heritable and results from the combination of genetic, neurologic, immunologic and environmental factors. However, it remains unclear whether its genetic component stems from the combination of a few common variants or of many rare variants.4, 5
Recent advances in genetics, genomics, developmental neurobiology and systems biology have offered important insights into the molecular agents and biological mechanisms responsible for ASD. Microarray technologies and next-generation sequencing have enabled high-throughput discovery of genes likely to be involved in the molecular pathology of autism.5, 6, 7, 8 However, as the success in discovery has risen, the number of candidate genes with associated risk for ASD has also stretched well into the hundreds.9, 10 As of December 2014, 667 genes have been implicated in autism (https://gene.sfari.org/autdb/HG_Home.do). Despite the large amounts of data now available, the general lack of replication across studies suggests that more data will be needed to fully characterize the genetic models responsible for the various forms of autism.
These high-throughput and large-scale efforts have confirmed that autism is a multisystem and heterogeneous condition. Thus, understanding the complex genetic architecture of ASD must involve, among other things, the study of autism gene expression across different tissues using integrative approaches. The majority of gene expression experiments conducted so far have been on blood-derived cells and to a lesser extent postmortem brain tissue from autism cases and matched controls. More recent approaches have examined regulatory patterns in induced pluripotent stem cells forming neurons from individuals with autism. Individually, these studies have advanced our understanding of molecular systems involved in either the cause or effect of autism. We propose and test here the notion that together these experiments may help refine our understanding of genes and pathways important in onset and maintenance of autism. Specifically, we perform an integrated systems biology analysis of all published autism gene expression studies to test whether a common signature representative of ASD exists and ultimately if it can be detected in both blood and brain.
Materials and methods
Experiments and gene lists
To compile a complete set of published and publically available gene expression experiments we used Nextbio,11 an ontology-based platform that provides global collections of high-throughput public data that meet four criteria: broad coverage of genes, existence of a control group, access to raw or normalized data and supply of sample annotations. We downloaded gene expression data and derived lists of differentially expressed genes from 27 case–control biosets of 9 independent experiments covering 657 autism, 9 mental retardation and developmental delay, 566 control samples: GSE37772, GSE25507, GSE7329, GSE28475, GSE38322, GSE6575, GSE18123, GSE28521 and GSE39447.7, 12, 13, 14, 15, 16, 17, 18, 19 Nextbio employed Welch or Standard t-test, paired and unpaired, as appropriate, to statistically analyze these case–control experiments and established a nominal, unadjusted P-value significance cutoff of 0.05 and a minimum absolute fold-change cutoff of 1.2 (the lowest sensitivity threshold of commercial microarray platforms) to select the differentially expressed genes for each bioset. The statistical threshold method used by Nextbio for gene selection aimed to maintain the balance between sensitivity and specificity across all experiments; these cutoffs were deliberately less stringent to guarantee the inclusion of all potentially interesting genes, and have been commonly used for this purpose in the literature.20, 21, 22, 23, 24, 25, 26 We then filtered each list of differentially expressed genes obtained from Nextbio to save just the unique gene symbol identities assigned from The Hugo Gene Nomenclature Comittee.27 A description and content of each bioset can be found in Supplementary Table S1a. It is important to clarify that, although important covariates exist across these experiments, age and gender were well-matched across a majority of the samples, with most of the mixed-gender biosets having roughly four males to one female (consistent with the male bias of autism). The studies whose data we used were careful to match the age and sex of their control groups to their case population (GSE18123, GSE28475, GSE38322, GSE6575 and GSE37772). In addition, studies with differences of age and/or gender between cases and control groups independently performed statistical analyses to confirm that neither age nor sex impacted the pattern of differential gene expression exhibited between control and case individuals (GSE28521, GSE25507); GSE7329 and GSE39447 studies examined only males, although matched for age. More information regarding number of subjects, age, gender, ethnicity and relationship among the individuals in each original study can be found in Supplementary Table S1b.
Cluster analysis
We then converted the transcriptional probe lists into a matrix of binary gene presence/absence with respect to each experimental case. We analyzed the matrix using the Jaccard coefficient in MATLAB (www.mathworks.com) to construct a gene-based dendrogram for all 27 biosets with the aim of determining the relatedness among them. This statistic measures the similarity and diversity among sample sets and is defined by the size of the intersection divided by the size of the union of sample sets. We elected to use this distance coefficient as it was originally developed for pattern discovery with binary matrices and does not treat the shared absence of a characteristic as evidence for relatedness, a valuable characteristic in the context of identifying similarity across gene expression experiments.
Cluster bootstrap validation
We utilized an integrated function of the ‘fpc’ package28 in R (http://www.R-project.org/), clusterboot(), for the assessment of clusterwise stability and validity of the clusters within the gene expression tree. We used a non-parametric bootstrapping procedure (B=1000 runs) to resample from the original data with replacement to construct bootstrap matrices and clusters, and iteratively used the Jaccard coefficient to compute the structural similarity of the resampled trees with the tree derived from the original data. We treated the mean of the Jaccard coefficients computed per permutation as the overall similarity between the original and permuted data, therefore an index of a cluster's stability. For each permutation, we set the paremeter k, the number of subsets, to 14 to match the total number of clusters obtained in the observed gene expression tree. Clusters supported by a Jaccard coefficient above 0.6 were treated as robust and stable, with values closer to 1.0 having the highest stability. Values ⩽0.5 were not stable and not considered in our analysis.
A more detailed explanation of the approach and justification for the cutoffs used here can be found in the study by Hennig.29 These cluster stability analyses were complemented with a classical multidimensional scaling approach that projects our dissimilarity data onto its first two principal dimensions, generated by the ‘showplots’ argument of Clusterboot() function. This resulted in a series of clusters including a collection of 66 genes that form the root of a statistically significant cluster involving both blood and brain experiments, and that we hereafter refer to as Root 66.
Gene annotation
To assess the biological context and potential significance of the Root 66 cluster, we used a manually curated database of genes linked to ASD, SFARI Gene30 and a systems medicine tool, Autworks,31 to extract information of autism candidate genes and candidate genes of related neuropsychiatric diseases together with an updated set of predicted gene interactions.
We also conducted a manual search for lists of variants that have been associated with autism candidate genes to increase coverage and reliability of the variant data used for validation of the biological significance of the Root 66 gene set.32, 33, 34, 35, 36
Functional and gene-network analyses
We performed biological pathway analysis using Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, www.ingenuity.com) to explore gene connectivity and related biological functions both within and across the disorders. IPA functional analysis associates biological functions and diseases with experimental results, including differentially expressed genes selected from microarray experiments. It leverages the biological interactions deposited in the manually curated Ingenuity Pathways Knowledge Base and organizes this information to provide statistical support for gene-to-gene associations. We imported corresponding Root 66 gene symbols into the Ingenuity Pathways Knowledge Base and generated networks using an edge rank score (P-score=−log10(P-value)) that indicated the likelihood of the genes co-occurring or interacting by random chance. A score >3 (P<0.001) suggested with more than 99.9% confidence that an edge between two genes was non-random. Finally, we carried out a ‘disease and function’ analysis to test whether the Root 66 genes had overrepresentation in specific human diseases and to explore the role(s) of the Root 66 genes in the context of statistically relevant biological processes pathways, and networks; IPA implemented a Fisher’s exact test to generate P-values to determine whether a biological process was enriched with genes of interest.
Comparison with other human disease gene expression
All RNA human expression disease vs normal experiments from Nextbio were manually curated for further analysis. In all, 1047 biosets were selected from 450 different experiments to perform pairwise intersections of the differentially expressed genes obtained in each experimental case–control comparison. We generated a matrix of the number of Root 66 genes present in each pairwise comparison using a custom Python script, and used RStudio37 to statistically explore the data sets and to build a distribution showing the frequency of Root 66 overlap for each bioset intersection.
Comparison with patterns of gene expression in normal tissue
We conducted a manual literature search to collect and annotate major gene expression studies38, 39, 40, 41, 42 of normal human tissues with the aim of investigating evidence of genes in the Root 66 gene set being involved in normal blood and/or brain transcriptional patterns. In addition, we assembled a manually curated list of 408 housekeeping genes from Reactome and KEGG pathway annotations43 to test whether Root 66 genes have previously been found to be widely expressed across normal tissue and involved in housekeeping roles.
Results
Comparative analysis of gene expression studies
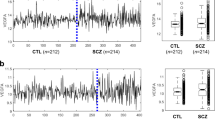
We generated lists of differentially expressed genes from ASD gene expression data analyses of 27 case–control biosets spanning 9 independent experiments: GSE37772, GSE25507, GSE7329, GSE28475, GSE38322, GSE6575, GSE18123, GSE28521 and GSE39447,7, 12, 13, 14, 15, 16, 17, 18, 19 available via Nextbio.11 Supplementary Table S1a provides a description of all biosets and Supplementary Table S1b contains information regarding number of subjects used in the experiments along with their age, gender and ethnicity. To determine relatedness across biosets, we converted the lists into a binary matrix of gene presence/absence and performed distance-based clustering with pairwise similarity measured via the Jaccard coefficient. As expected, the majority of the 27 biosets clustered together by experiment first and tissue type second (Figure 1, also Supplementary Figure S1), suggesting that the likely cause of the significant clustering of these biosets was variation related to the experimental design and nature of the samples. However, one subgroup of the tree deviated from this expectation and included biosets from three different experiments (GSE18123, GSE38322 and GSE28475) involving both brain and blood tissue (Figure 1). This subgroup was rooted by a collection of 66 genes (see Table 1 and Supplementary Table S2) and was statistically supported by bootstrap resampling (see Figure 2, also Supplementary Figure S2 and Supplementary Table S3). It is important to note that this Root 66 cluster was the only one that achieved statistical significance that integrated 3 different experiments from different tissues (brain and blood) across 3 independent expression platforms (GPL570 from Affymetrix (Santa Clara, CA, USA) and GPL6883 and GPL10558 from Illumina (Hayward, CA, USA)) and a diversity of subjects included (ASD and control males and females with different ages). This suggested that the source of the clustering was not co-variables but instead the regulatory patterns represented by these 66 genes.

Gene-based clustering of the 27 biosets (see Supplementary Table S1). The majority clustered together by experiment first and tissue type second, with the exception of the Root 66 subgroup (highlighted in purple).

Jaccard clustering of the 27 biosets generated by bootstrap with replacement (k=14). The Root 66 subgroup, highlighted in purple, presented a stability index value of 0.617, suggesting that it was a non-random group of probable biological significance.
To evaluate the hypothesis that this Root 66 could represent a common signature in blood and brain indicative of autism, we examined the genetic network formed by these genes, their implication in other autism-related neurological conditions and their mutational burden as reported by recently published exome-sequencing studies.32, 33, 34, 35, 36
Links to autism
Four genes in our candidate list for autism (MAP1LC3B, PDE4B, TCF4 and UPF2) have already been associated with ASD. In addition, 32 interact directly with genes that have been associated with autistic disorder. An additional 19 are directly involved in or have links with genes associated with related neurological conditions, including schizophrenia, epilepsy, intellectual disability, seizures, attention deficit and disrupted behavior disorders, Angelman syndrome, bipolar disorder, mental retardation, developmental disabilities, sleep disorders and Alzheimer's disease, among others (Table 1). Supplementary Table S2 details the Root 66 genes including links to neurological disorders and interaction with known autism genes. Triangulating with recently published exome-sequencing studies, nine of the Root 66 genes have had variants reported as de novo or elevated risk for autism,32, 33, 34, 35, 36 and Table 2.
Pathway enrichment
Using IPA, we determined that the Root 66 gene set was enriched, with IPA edge rank scores >3, in a total of 3 biological networks related to neurological function. These are detailed in the three sections below:
Neuroendocrine and normal development network (IPA score=27, P≪0.01)
The first network (Supplementary Figure S3) included 15 Root 66 genes, 2 of them already associated with autism (MAP1LC3B and PDE4B), that interact in neurological processes involved in normal brain growth and function, such as formation of astrocytes, proliferation of cortical neurons, sorting of axons and myelination. Dysregulation of Root 66 genes may affect relevant brain processes such as learning and memory, as they were found to be linked to relevant nodes in the network (PI3K and NFKB complexes) that are implicated in postsynaptic density and glutamatergic synapses, and, therefore, in synaptic plasticity. This network also showed an interesting connection between genes associated with ASD and other related neurological disorders, and endocrine hormones of the hypothalamic–pituitary–gonadal axis, such as follicle-stimulating hormone, luteinizing hormone and gonadotropin-releasing hormone.
Neurodegeneration network (IPA score=24, P≪0.01)
A second network (Supplementary Figure S4) included 14 Root 66 genes (UPF2 already linked to autism) that have been shown to interact with molecules involved in mechanisms responsible for nervous system inflammation, loss of neurological function and abnormal morphology of the brain. Several genes central in the network, such as APP, PTGS2, ERG and YWHAG were found to be linked to Root 66 genes, and have been implicated in processes such as amyloid plaque formation, astrocytosis and gliosis, cognitive degeneration and neurological dysfunction typical of neurological diseases including Parkinson's, Alzheimer's and Schizophrenia.
Neurodegeneration and tumor network (IPA score=22, P≪0.01)
Thirteen Root 66 genes were found to be interacting in this network (Supplementary Figure S5). Three genes involved in cancer appeared to be the most connected nodes: TP53, PTEN and AGTR1. The network was enriched in biological processes such as neurodegeneration, abnormal morphology and damage of the nervous system, likely caused by tumorigenesis.
While the above 3 networks showed potentially independent roles of smaller sets of genes contained with the Root 66, we also queried the global connectivity among member genes of Root 66. To do so, we merged the three networks into a single global gene network (Figure 3). The merged network connected 42 of the Root 66 genes (Figure 3) and revealed direct links between these Root 66 genes and genes participating in pathways related to neurological processes, such as synaptic transmission, neurodegeneration, learning and memory, as described above in the independent networks and labeled in Supplementary Figures as provided by IPA. Several autism-related diseases and functions were statistically enriched in this global network; gene names and corresponding P-values can be found in Table 3. We found evidence to support the role of the Root 66 network in biological mechanisms that cause alteration of the nervous system, and, therefore, neurological disorders, and also involvement in morphologic and molecular alterations of normal brain development that may underlie the etiology of ASD. While this global network was statistically significant, more testing will be required to confirm its biological significance and potential role in autism.

Biological network formed by the Root 66 gene set. Forty-two Root 66 genes (highlighted in purple) are tightly connected and interact in biological processes related to neurological conditions indicated in synaptic transmission, neurodegeneration, abnormal brain morphology, and learning and memory (Table 3). Interactions with any of the Root 66 genes are highlighted in blue.
Root 66 expression in non-autism disease and normal tissue
We manually selected 1047 biosets from Nextbio11 containing genes differentially expressed between disease and normal tissue from 450 independent RNA expression experiments. These experiments contained a diversity of tissue types roughly comparable to the experiments used in our primary analysis. We then computed pairwise intersections between these biosets to assess the frequency of appearance of our Root 66 in each overlap. The distribution displayed in Supplementary Figure S6 details the number of Root 66 genes found in each pairwise bioset intersection. The overlap was negligible and not statistically significant. The average number of genes found in the intersection among these biosets was 6.15 compared with the 66 genes obtained in individuals with autism. In addition, we found five relevant studies38, 39, 40, 41, 42 where hierarchical clustering of normal tissues was made using different approaches; none of these showed a grouping between brain and blood samples. Instead, these two tissue types segregated separately into unconnected branches within the dendrogram. In addition, only 3 Root 66 genes (RBMX, SUPT4H1 and UBE2D3) intersected with our list of 408 experimentally confirmed housekeeping genes.43 These findings collectively lend support to the hypothesis that the genes in the Root 66 cluster play unique roles in autism.
Discussion
In this study, we compared a large set of published and openly available gene expression experiments from different tissue types performed in individuals with autism, with the goal of testing the hypothesis that a signature of autism can be found in the blood that might be a molecular echo of autism-related regulatory impairment in the brain.
We discovered a statistically significant subgroup that deviated from expectation and included biosets from three independent experiments involving both brain and blood tissue. This subgroup was significantly statistically supported by a subset of 66 genes that we termed Root 66. Four of the Root 66 genes (MAP1LC3B, PDE4B, TCF4 and UPF2) have previously been associated with autism, and 56 either have been shown to interact directly with known autism candidates or have been implicated in other autism-related neurological disorders.
To better understand the biological significance of Root 66, we tested its member genes’ enrichment in specific biological processes and whether these genes form an interconnected biological network. Our analysis revealed that a significant number of Root 66 genes were interacting in 3 relevant networks, each of them related to neurological disease in a different but potentially significant way. The first network was enriched in biological processes related to brain growth and development that may affect learning and memory, and also showed some dysregulation in neuroendocrine activity, particularly an interaction between Root 66 genes and endocrine hormones of the hypothalamic—pituitary—gonadal axis. Although the main role of this axis is to control development, reproduction and aging, it is known that these hormones affect behavior, as they have been shown to alter brain structure and functioning including dsyregulation of the follicle-stimulating hormone, which has known roles in brain development and neuronal differentiation.44 Moreover, the activity of gonadotropin-releasing hormone neurons, and thus, the regulation of gonadotropin release in blood, is stimulated or inhibited by oxytocin, a neurohypophysial hormone that also acts as a neurotransmitter in the brain. Several studies45, 46, 47, 48, 49 have revealed that oxytocin is implicated in social behavior, recognition and bonding, as well as the establishment of trust among people. Furthermore, there is evidence that alterations in the neuromodulatory role of oxytocin are linked to a variety of mental disorders, including autism.50, 51, 52 Finally, polymorphisms in the gene that codes for the oxytocin receptor have been associated with ASD risk.53
The second network showed Root 66 gene enrichment in nervous system inflammation, loss of neurological function and abnormal morphology of brain, supporting roles in neurodegeneration. There is evidence54, 55 of neural cell loss and activation of microglia and astrocytes in ASD, as well as high levels of APP.56, 57, 58 These studies suggested that neurodegeneration may have a role in autism, as it could explain the loss of previously acquired skills and abilities indicative of regressive forms of autism. Alterations in common neurological mechanisms, such as disruption during synaptogenesis may relate to ASD and other disorders of the brain, including schizophrenia, epilepsy, Alzheimer's disease and Parkinson's disease. For example, work has shown evidence for impaired neural synchrony and neurotransmission systems as pathophysiological processes involved in onset and/or maintenance of these neurological conditions.59, 60, 61, 62, 63, 64
Finally, the third network was enriched in neurodegeneration, abnormal morphology and damage of the nervous system and included the known cancer genes TP53, PTEN and AGTR1 as the top most connected nodes. Mutations in tumor suppressor gene PTEN, known to be associated with thyroid, breast and colon cancers, have been found in subgroups of children with autism who also have comorbid conditions of macrocephaly and/or epilepsy.65, 66 In addition, it has been suggested that PTEN mutations can have downstream impacts on other autism gene candidates, perhaps playing a role in the autism phenotype.67 Work has shown that when defective PTEN interacts with TP53 a decrease in the energy production of neurons occurs, leading to stress that induces mitochondrial DNA changes and abnormal levels of energy production in brain regions that are crucial for social behavior and cognition.68
Merging of these networks revealed that Root 66 genes formed a tightly connected network enriched in neurological processes such as synaptic transmission, neurodegeneration, abnormal brain morphology, learning and memory, supporting its potential role in neurological impairment, and in particular, autism. We next ran several tests to determine whether Root 66 represented a unique signature in autism unlikely to occur by chance or to represent a more ubiquitous signature of neurological impairment. We explored the list of differentially expressed genes in 450 disease vs normal RNA expression experiments for overlap with Root 66. The mean overlap of 6 genes strongly supported the hypothesis that the brain–blood cluster formed by Root 66 was unlikely to be a chance event and may represent a pattern unique to autism.
To further test whether the Root 66 brain–blood cluster observed in our study could form by chance, we explored the results from 5 studies38, 39, 40, 41, 42 of normal brain and blood gene expression. These studies showed that although there is overlap in the genes expressed in normal brain and blood, the different tissue transcriptomes clustered independently from one another in all cases, unlike what we observed in the present analysis. We also tested whether the genes in Root 66 played more generic roles as housekeeping genes.69 Only 3 of its member genes overlapped with an experimentally confirmed list of 408 housekeeping genes manually curated from Reactome and KEGG pathway databases.43 Both results lend additional support to the hypothesis that the Root 66 cluster is non-random and likely plays a role unique to autism.
Conclusion
Gene expression studies published to date have had a relatively limited impact on our understanding of autism’s molecular pathology. Here we show by integrating and analyzing several published gene expression experiments a statistically significant signal between blood and brain rooted by 66 genes. The Root 66 gene set appeared to be non-random and of potential etiologic relevance to autism, as most of its members have known association with neurological processes crucial for normal brain development and function and confirmed roles in neurological disease. While further independent replication and experimental validation will be needed to confirm our preliminary findings, the current results suggest that there is a detectable signature in the blood of individuals with autism that echoes what might be an important signature of dysregulation in the brain.
References
Centers for Disease Control and Prevention. Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators. Prevalence of autism spectrum disorders—autism and developmental disabilities monitoring network, 14 Sites, United States, 2008. MMWR Morb Mortal Wkly Rep 2012; 61: 1–19.
Vardarajan BN, Eran A, Jung J-Y, Kunkel LM, Wall DP . Haplotype structure enables prioritization of common markers and candidate genes in autism spectrum disorders. Transl Psychiatry 2013; 3: e262.
Lai MC, Lombardo MV, Baron-Cohen S . Autism. Lancet 2014; 383: 896–910.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T et al. Strong association of De Novo copy number mutations with autism. Science 2007; 316: 445–449.
Geschwind DH, Jeste SS . Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat Rev Neurol 2014; 10: 74–81.
Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet 2013; 93: 249–263.
Luo R, Sanders SJ, Tian Y, Voineagu I, Huang N, Chu SH et al. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am J Hum Genet 2012; 91: 38–55.
Matsunami N, Hensel CH, Baird L, Stevens J, Otterud B, Leppert T et al. Identification of rare DNA sequence variants in high-risk autism families and their prevalence in a large case/control population. Mol Autism 2014; 5: 5.
Abrahams BS, Geschwind DH . Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet 2008; 9: 341–355.
Freitag CM . The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry 2007; 12: 2–22.
Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J et al. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One 2010; 5: e13066.
Alter MD, Kharkar R, Ramsey KE, Craig DW, Melmed RD, Grebe TA et al. Autism and increased paternal age related changes in global levels of gene expression regulation. PLoS One 2011; 6: e16715.
Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M et al. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet 2007; 16: 1682–1698.
Chow ML, Winn ME, Li HR, April C, Wynshaw-Boris A, Fan JB et al. Preprocessing and quality control strategies for Illumina DASL assay-based brain gene expression studies with semi-degraded samples. Front Genet 2012; 3: 11.
Ginsberg MR, Rubin RA, Falcone T, Ting AH, Natowicz MR . Brain transcriptional and epigenetic associations with autism. PLoS One 2012; 7: e44736.
Gregg JP, Lit L, Baron CA, Hertz-Picciotto I, Walker W, Davis RA et al. Gene expression changes in children with autism. Genomics 2008; 91: 22–29.
Kong SW, Collins CD, Shimizu-Motohashi Y, Holm IA, Campbell MG, Lee IH et al. Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PLoS One 2012; 7: e49475.
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474: 380–384.
Novarino G, El-Fishawy P, Kayserili H, Meguid NA, Scott EM, Schroth J et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 2012; 338: 394–397.
Fan X, Shi L, Fang H, Harris S, Perkins R, Tong W . Investigation of reproducibility of differentially expressed genes in DNA microarrays through statistical simulation. BMC Proc 2009; 3: S4.
Shi L, Jones WD, Jensen RV, Harris SC, Perkins RG, Goodsaid FM et al. The balance of reproducibility, sensitivity, and specificity of lists of differentially expressed genes in microarray studies. BMC Bioinformatics 2008; 9: S10.
Grigoryev DN, Ma SF, Irizarry RA, Ye SQ, Quackenbush J, Garcia JG . Orthologous gene-expression profiling in multi-species models: search for candidate genes. Genome Biol 2004; 5: R34.
Knowles LM, Smith JW . Genome-wide changes accompanying knockdown of fatty acid synthase in breast cancer. BMC Genomics 2007; 8: 168.
Li Y, Elashoff D, Oh M, Sinha U St, John MA, Zhou X et al. Serum circulating human mRNA profiling and its utility for oral cancer detection. J Clin Oncol 2006; 24: 1754–1760.
Wang Y, Barbacioru C, Hyland F, Xiao W, Hunkapiller KL, Blake J et al. Large scale real-time PCR validation on gene expression measurements from two commercial long-oligonucleotide microarrays. BMC Genomics 2006; 7: 59.
Wen Z, Su Z, Liu J, Ning B, Guo L, Tong W. The MicroArray Quality Control (MAQC) Project and Cross-Platform Analysis of Microarray Data. In: Horng-Shing Lu H, Schölkopf B, Zhao H (eds). Handbook of Statistical Bioinformatics. Springer Berlin Heidelberg: Berlin, Germany, 2011, pp 171–192..
Gray KA, Yates B, Seal RL, Wright MW, Bruford EA . Genenames.org: the HGNC resources in 2015. Nucleic Acids Res 2015; 43: D1079–D1085.
Hennig C fpc: Flexible procedures for clusteringR package Version 2.1–7. http://CRAN.R-project.org/package=fpc, 2014.
Hennig C . Cluster-wise assessment of cluster stability. Comput Stat Data Anal 2007; 52: 258–271.
Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism 2013; 4: 36.
Nelson TH, Jung JY, Deluca TF, Hinebaugh BK St, Gabriel KC, Wall DP . Autworks: a cross-disease network biology application for autism and related disorders. BMC Med Genomics 2012; 5: 56.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012; 74: 285–299.
Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012; 485: 242–245.
O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012; 338: 1619–1622.
O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012; 485: 246–250.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012; 485: 237–241.
Racine JS . RStudio: a platform-independent IDE for R and Sweave. J Appl Econ 2012; 27: 167–172.
Shyamsundar R, Kim YH, Higgins JP, Montgomery K, Jorden M, Sethuraman A et al. A DNA microarray survey of gene expression in normal human tissues. Genome Biol 2005; 6: R22.
Jongeneel CV, Delorenzi M, Iseli C, Zhou D, Haudenschild CD, Khrebtukova I et al. An atlas of human gene expression from massively parallel signature sequencing (MPSS). Genome Res 2005; 7: 1007–1014.
Dezső Z, Nikolsky Y, Sviridov E, Shi W, Serebriyskaya T, Dosymbekov D et al. A comprehensive functional analysis of tissue specificity of human gene expression. BMC Biol 2008; 6: 49.
Hsiao LL, Dangond F, Yoshida T, Hong R, Jensen RV, Misra J et al. A compendium of gene expression in normal human tissues. Physiol Genomics 2001; 7: 97–104.
Chang CW, Cheng WC, Chen CR, Shu WY, Tsai ML, Huang CL et al. Identification of human housekeeping genes and tissue-selective genes by microarray meta-analysis. PLoS One 2011; 6: e22859.
Zhu J, He F, Song S, Wang J, Yu J . How many human genes can be defined as housekeeping with current expression data? BMC Genomics 2008; 9: 172.
Vadakkadath Meethal S, Atwood CS . The role of hypothalamic-pituitary-gonadal hormones in the normal structure and functioning of the brain. Cell Mol Life Sci 2005; 62: 257–270.
Kosfeld M, Heinrichs M, Zak PJ, Fischbacher U, Fehr E . Oxytocin increases trust in humans. Nature 2005; 435: 673–676.
Baumgartner T, Heinrichs M, Vonlanthen A, Fischbacher U, Fehr E . Oxytocin shapes the neural circuitry of trust and trust adaptation in humans. Neuron 2008; 58: 639–650.
Wittfoth-Schardt D, Gründing J, Wittfoth M, Lanfermann H, Heinrichs M, Domes G et al. Oxytocin modulates neural reactivity to children's faces as a function of social salience. Neuropsychopharmacology 2012; 37: 1799–1807.
Ross HE, Young LJ . Oxytocin and the neural mechanisms regulating social cognition and affiliative behavior. Front Neuroendocrinol 2009; 4: 534–547.
Peñagarikano O, Lázaro MT, Lu XH, Gordon A, Dong H, Lam HA et al. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci Transl Med 2015; 7: 271ra8.
Jacobson JD, Ellerbeck KA, Kelly KA, Fleming KK, Jamison TR, Coffey CW et al. Evidence for alterations in stimulatory G proteins and oxytocin levels in children with autism. Psychoneuroendocrinology 2014; 40: 159–169.
Marazziti D, Catena Dell'osso M . The role of oxytocin in neuropsychiatric disorders. Curr Med Chem 2008; 15: 698–704.
Gordon I, Vander Wyk BC, Bennett RH, Cordeaux C, Lucas MV, Eilbott JA et al. Oxytocin enhances brain function in children with autism. Proc Natl Acad Sci USA 2013; 110: 20953–20958.
Damiano CR, Aloi J, Dunlap K, Burrus CJ, Mosner MG, Kozink RV et al. Association between the oxytocin receptor (OXTR) gene and mesolimbic responses to rewards. Mol Autism 2014; 5: 7.
Kern JK, Geier DA, Sykes LK, Geier MR . Evidence of neurodegeneration in autism spectrum disorder. Transl Neurodegener 2013; 2: 17.
Edmonson C, Ziats MN, Rennert OM . Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol Autism 2014; 5: 3.
Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA et al. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J Child Neurol 2006; 21: 444–449.
Ray B, Long JM, Sokol DK, Lahiri DK . Increased secreted amyloid precursor protein-α (sAPPα) in severe autism: proposal of a specific, anabolic pathway and putative biomarker. PLoS One 2011; 6: e20405.
Lahiri DK, Sokol DK, Erickson C, Ray B, Ho CY, Maloney B . Autism as early neurodevelopmental disorder: evidence for an sAPPα-mediated anabolic pathway. Front Cell Neurosci 2013; 7: 94.
Uhlhaas PJ, Singer W . Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron 2006; 52: 155–168.
Meyer U, Feldon J, Dammann O . Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res 2011; 69: 26R–33R.
de Lacy N, King BH . Revisiting the relationship between autism and schizophrenia: toward an integrated neurobiology. Annu Rev Clin Psychol 2013; 9: 555–587.
Sokol DK, Maloney B, Long JM, Ray B, Lahiri DK . Autism, Alzheimer disease, and fragile X. APP, FMRP, and mGluR5 are molecular links. Neurology 2011; 76: 1344–1352.
Fujita-Jimbo E, Yu Z-L, Li H, Yamagata T, Mori M, Momoi T et al. Mutation in Parkinson disease-associated, G-protein-coupled receptor 37 (GPR37/PaelR) is related to autism spectrum disorder. PLoS One 2012; 7: e51155.
Hollander E, Wang AT, Braun A, Marsh L . Neurological considerations: autism and Parkinson's disease. Psychiatry Res 2009; 170: 43–51.
Marchese M, Conti V, Valvo G, Moro F, Muratori F, Tancredi R et al. Autism-epilepsy phenotype with macrocephaly suggests PTEN, but not GLIALCAM, genetic screening. BMC Med Genet 2014; 15: 26.
Varga EA, Pastore M, Prior T, Herman GE, McBride KL . The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med 2009; 11: 111–117.
Zhou J, Parada LF . PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol 2012; 22: 873–879.
Napoli E, Ross-Inta C, Wong S, Hung C, Fujisawa Y, Sakaguchi D et al. Mitochondrial dysfunction in Pten haplo-insufficient mice with social deficits and repetitive behavior: interplay between Pten and p53. PLoS One 2012; 7: e42504.
Butte AJ, Dzau VJ, Glueck SB . Further defining housekeeping or ‘maintenance’ genes. Focus on ‘A compendium of gene expression in normal human tissues’. Physiol Genomics 2001; 7: 95–96.
Acknowledgements
We are grateful to all the members of the DPW Lab, specially Maude David and Chloe O’Connell for helpful discussions, their time and assistance. In addition, we thank the reviewers and the editor whose valuable comments substantially improved the quality of the manuscript. We also would like to thank all the families who contributed the data that made this study possible. This work was supported by funds to DPW from grant 1R01MH090611 from the National Institutes of Health and the Hartwell Foundation's Autism Research and Technology Initiative (iHART).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Diaz-Beltran, L., Esteban, F. & Wall, D. A common molecular signature in ASD gene expression: following Root 66 to autism. Transl Psychiatry 6, e705 (2016). https://doi.org/10.1038/tp.2015.112
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2015.112
This article is cited by
-
A predictive ensemble classifier for the gene expression diagnosis of ASD at ages 1 to 4 years
Molecular Psychiatry (2023)
-
Game theoretic centrality: a novel approach to prioritize disease candidate genes by combining biological networks with the Shapley value
BMC Bioinformatics (2020)
-
RETRACTED ARTICLE: Regional patterning of co-expressed genes in autistic brains
Network Modeling Analysis in Health Informatics and Bioinformatics (2019)
-
Brain-specific functional relationship networks inform autism spectrum disorder gene prediction
Translational Psychiatry (2018)
-
PTSD Blood Transcriptome Mega-Analysis: Shared Inflammatory Pathways across Biological Sex and Modes of Trauma
Neuropsychopharmacology (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}