
Abstract
Some individuals suffering from posttraumatic stress disorder (PTSD) exhibit lower basal salivary cortisol and higher glucocorticoid receptor (GR) sensitivity. Recent studies suggest that epigenetic mechanisms regulate the activity of cortisol and GR. As a means to combine and cross-validate those findings, we compared cortisol, GR expression and promoter methylation levels in peripheral T lymphocytes of healthy controls versus individuals endorsing a diagnosis of lifetime PTSD. Thirty subjects with lifetime (current or remitted) PTSD and 16 subjects never exposed to trauma were recruited. Salivary cortisol was collected at six time points over the course of a single weekday and analyzed utilizing a time-resolved fluorescence immunoassay. GR expression (GRtotal, 1B, 1C, 1F and 1H) was measured by quantitative RT-PCR. DNA methylation levels in human glucocorticoid receptor (hGR) 1B and 1C variant’s promoter were quantified by epityper in T lymphocytes isolated by magnetic-assisted cell sorting. Individuals with lifetime PTSD have lower morning cortisol release, higher mRNA expression of hGRtotal, 1B, and 1C and lower overall methylation levels in hGR 1B and 1C promoters. Cortisol levels were inversely correlated with hGR 1B mRNA expression. Moreover, overall and CpG site-specific methylation levels were inversely correlated with hGRtotal and 1B mRNA expression. There was no difference between current and remitted PTSD across cortisol, GR expression mRNA and DNA methylation data. Traumatic events induce DNA methylation alterations in distinct promoters of hGR with transcriptional modifications that associate with hypoactive hypothalamus-pituitary-adrenal axis in individuals with PTSD. Our results also point toward an important role of hGR 1B variant in PTSD.
Similar content being viewed by others


Introduction
Posttraumatic stress disorder (PTSD) is characterized by symptoms of intrusions, avoidance and hyperarousal following exposure to a traumatic event. Lifetime trauma exposure has been estimated between 40–90%.1, 2, 3, 4, 5, 6 However, the lifetime prevalence for PTSD is 7–12%.1,7
If remission is considered the normative response to trauma exposure, a substantial proportion of individuals go on developing long-lasting intractable PTSD. The clinical manifestations of PTSD may reflect enduring changes in the stress-related neurobiological structures and systems.8 Indeed, changes in the activity of the hypothalamus-pituitary-adrenal (HPA) axis, the main stress regulatory system, are induced by exposure to severe stressors in animals and humans9,10 and dysfunction of the HPA axis activity is a well-characterized feature in PTSD. For instance, such patients exhibit higher corticotropin releasing factor in the cerebrospinal fluid,11,12 lower plasma and serum morning cortisol release,13 higher glucocorticoid receptor (GR) expression in lymphocytes14 and greater GR sensitivity.15 In addition, recent studies have shown that life-events, at least early in life, can modify the chemical structure surrounding the genome and influence gene expression,16 including GR expression.17, 18, 19
The human GR gene contains eight coding exons (exons 2–9) and nine 5’ non-coding first exons (exons A, B, C, D, E, F, H, I, J) spanned through a region of more than 80 kb within chromosome 5q31-q32.20, 21, 22, 23 Both rodent and human GR levels are regulated almost exclusively at the transcriptional level24,25 and each non-coding exon 1 variant has its own promoter.22,26, 27, 28, 29, 30 These multiple independent promoter regions contribute to the tissue specificity of GR expression.23 DNA methylation is one of the epigenetic mechanisms controlling the expression of genes and has been shown to be modified by environmental stress.31 For instance, in rats, maternal behavior regulates hippocampal GR17 expression in offspring through changes in GR17 promoter methylation.17 Similarly, decreased hippocampal total GR, 1B, 1C, 1F (rat homolog for human GR1F) and 1H expression associates with respective GR exon 1 variant promoter methylation alterations in suicide completers who were abused in childhood.18,19 These results suggest that methylation changes in GR promoters modify GR expression in individuals who experienced early-life adversity. It remains unknown if epigenetic regulation also explains HPA axis activity in other conditions associated with adversity such as PTSD. However, by definition, PTSD is etiologically related to environmental stress and, as such, it is likely to involve an important epigenetic component.
To address this question, we examined HPA axis functioning in individuals endorsing a diagnosis of lifetime PTSD by investigating salivary cortisol measures, the expression of four variants of GR (1B, 1C, 1F and 1H), and their association with variations in exon 1 variant promoter DNA methylation in T lymphocytes. We investigated T lymphocytes because these cells have an important role in cell-mediated immunity, and as such, are depositaries of cellular memory to environmental factors. In addition, DNA methylation patterns vary between different cell fractions,32 thus it is preferable to investigate DNA extracted from individual cell populations rather than from homogenates. We hypothesized that the HPA axis alterations in PTSD are associated with variations in cortisol levels due to changes in the expression of the GR gene. We further hypothesized that modifications in DNA methylation levels within the promoter region of the GR gene are associated with changes in GR expression in PTSD.
Materials and methods
Subjects and procedure
Twenty seven individuals meeting DSM-IV criteria for lifetime PTSD (Current PTSD: N=15; Past PTSD: N=12) were recruited. Diagnosis was established with a semi-structured interview33 and further corroborated by a self-report symptom measure.34 Sixteen individuals with a negative trauma history were also recruited. Current comorbid depressive symptoms were assessed with a subscale of the symptom check list-90 using a cutoff of 70 for clinically relevant symptoms of depression.35 The index traumatic events were as follows: motor vehicle accident (n=19), participation in a peacekeeping mission (n=1), assault with a weapon (n=2), physical and/or sexual abuse in adulthood (n=14), physical and/or sexual abuse during childhood (n=13) or other (n=8). All events met the DSM-IV A1 (life threat) and A2 (fear, helplessness or horror) criteria for trauma exposure. Exclusion criteria for controls were a lifetime trauma exposure, a current DSM-IV axis I psychiatric diagnosis36 including any form of substance abuse and living outside of the greater Montreal metropolitan area. Table 1 lists sociodemographic and clinical sample’s information. The Research Ethics Board at the Douglas University Mental Health Institute approved the project. Signed informed consent was obtained from every participant.
Salivary cortisol
Basal salivary cortisol (unbound free cortisol) was sampled following previously described methods.37,38 Participants were given six saliva collection tubes (Sarstedt, Montreal, QC, Canada ) to use at home over the course of a single weekday at the following times: awakening, awakening+30 min, awakening+45 min, awakening+60 min, awakening+8 h and bedtime. The saliva samples were stored at −80 °C. Samples were analyzed in duplicate all at once using a time-resolved fluorescence immunoassay.38 The average coefficient of variation between duplicates was 5.2%. Cortisol data were plotted on a graph and the area under the curve was measured for each time interval according to previously described methods.39
Peripheral blood RNA extraction
Blood samples drawn for the RNA extraction (12 ml) by a registered nurse were immediately put on Leucolock (total RNA isolation system, Ambion, CA, USA) and frozen at −80 °C until RNA extraction the next day. As opposed to DNA (see next section), RNA was extracted from whole blood. Once extracted, concentrations and quality were measured using a 2100 BioAnalyzer (Agilent Technologies, Palo Alto, CA, USA). Samples with a RNA Integrity Number greater than 5.5 on a scale of 1 (lowest) to 10 (highest) were used for further analysis.
Human glucocorticoid receptor expression analysis
RNA was converted to cDNA using oligo dT primers according to manufacturer’s recommendations (Invitrogen, Burlington, ON, Canada) and expression was analyzed using a real time polymerase chain reaction (RT-PCR) in an ABI 7900HT (Applied Biosystems, Foster City, CA, USA). Amplification of cDNA was performed using custom-designed taqman probes used in a previous study (Supplementary Table S1). Primers and taqman probes were obtained from Integrated DNA Technology (San Diego, CA, USA; Supplementary Table S1). Quantification was performed with five replicates each in a 12 μl reaction as followed: 0.6 μl of GR primers/probes, 3.1 μl of water, 6.0 μl of Taqman Master Mix and 2 μl of cDNA (1 ng μl−1). Thermal cycling conditions were: 50 °C for 2 min, 95 °C for 10 min and 40 cycles, each of 95 °C for 15 s and specific melting temperature for 60 s (Supplementary Table S1). The cycle threshold values for all replicates were pooled to obtain a mean value. Replicates with a standard deviation greater than 0.3 were excluded from the analysis. For each replicate, a quantity was extrapolated from a standard curve including six dots, each of which was five times diluted (5:1). A standard curve was composed of a mix cDNA from all subjects. Mean quantities from all sample replicates were normalized to each of the reference genes (β-Actin, GAPDH and 18S) and averaged together. Expression data are expressed as ratios of average mean quantities.
T-lymphocyte isolation, DNA extraction and methylation quantification
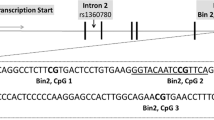
Blood samples drawn for the T-lymphocyte isolation and DNA extraction (12 ml) were immediately put on ice before being processed on the same day. T lymphocytes were isolated following the Automacs Pro protocol using CD3 whole blood microbeads according to manufacturer’s recommendations (Miltenyi Biotech, Boston, MA, USA). T-lymphocyte DNA was extracted using the DNA micro kit (Qiagen, Toronto, ON, Canada). DNA was diluted to 500 ng and sodium bisulfite conversion was performed using the EpiTect Bisulfite kit (Qiagen) according to the manufacturer’s instructions. Bisulfite-treated DNA was then sent to the Innovation Center of Genome Quebec (Montreal, Quebec, Canada) where epityper (Sequenom, San Diego, CA, USA) was performed on the regions of human glucocorticoid receptor (hGR) promoters defined in Supplementary Figure S3. Methylation data are expressed as mean percentage of overall methylation (global methylation) and methylation at each CpG site.
Statistical analyses
Analyses were conducted using SPSS 19.0 (IBM Corp., Armonk, NY, USA). Analyses were adjusted for age and sex. Gene expression values were normalized and compared using univariate analysis of variance (ANOVA). Cortisol and methylation levels were analyzed using a mixed model analysis with group (PTSD and controls) as a fixed factor and cortisol sampling time (six samples) or CpG sites as a repeated measure, followed by LSD post hoc tests. For each analysis, outliers within groups were excluded when their value differed by more than two SDs from the group mean (Z-test). Correlations between cortisol, expression and methylation levels were analyzed with Pearson’s correlation. Statistical significance was fixed at P<0.05. Data are presented as mean±standard error of the mean (s.e.m.).
Results
Salivary basal cortisol levels
A mixed two-way ANOVA performed on overall cortisol levels (area under the curve) revealed a significant main effect of group (F(1,87)=15.2, P<0.0005), with cortisol levels being lower in the PTSD group compared with controls (Figure 1). The analysis also revealed a main effect of cortisol sampling time (F(4,40)=151.1, P<0.0001) and a group by time interaction (F(4,40)=4.6, P<0.005). Post hoc tests indicated that cortisol levels were significantly lower in the PTSD group compared with the controls for the time interval between 30 and 45 min post awakening (P<0.01) and between 45–60 min post awakening (P<0.05; Figure 1). In addition, a between-groups trend was found for the time interval between awakening and 30 min post awakening (P=0.058). An analysis of covariance (ANCOVA) revealed no between-group effect of age, sex and depression on cortisol levels.

Diurnal variations in basal salivary cortisol levels (mean±s.e.m.) at awakening, +30 min, +45 min, +60 min, 4pm and bedtime as a function of group status. Note that time intervals are plotted evenly. Lifetime PTSD: n=22, Controls: n=15. *P<0.05; †P=0.058. CTRL, control; PTSD, posttraumatic stress disorder.
We also compared cortisol levels in current and past PTSD patients. Interestingly, we found no significant difference in the cortisol levels between current and past PTSD (F(4,23)=0.106, P=0.98) suggesting that changes in the cortisol levels are maintained after remission from PTSD symptoms (Supplementary Figure S1A). These effects were also independent of age and sex.
Whole blood human glucocorticoid receptor expression levels
The relative expression of hGRtotal as well as the first non-coding exons 1B, 1C, 1F and 1H was compared across the PTSD and control groups in peripheral blood samples. Results showed a significantly higher expression of hGRtotal (F(1,43)=5.3; P<0.05; Figure 2a), 1B (F(1,43)=12.1; P<0.005; Figure 2b) and 1C (F(1,42)=4.2; P<0.05; Figure 2c) in the PTSD group compared with controls. In contrast, hGR 1H relative expression was not significantly different between the groups (Figure 2d). In addition, consistent with previous findings showing that hGR 1F expression is mainly restricted to the brain,23 we were unable to quantify its expression in peripheral blood. ANCOVA revealed no effect of age or sex on hGRtotal, 1B and 1C expression levels between groups although we found a significant effect of age on hGR 1H expression.

Glucocorticoid receptor mRNA expression levels (average relative mean quantity±s.e.m.) of (a) hGRtotal, (b) hGR 1B, (c) hGR 1C, and (d) hGR 1H as a function of group status. PTSD: n=27, Controls: n=16. *P<0.05. CTRL, control; hGR, human glucocorticoid receptor; PTSD, posttraumatic stress disorder.
Interestingly, we found a significant negative correlation between hGR 1B relative expression and cortisol levels for the time intervals between awakening and 30 min post awakening (r=−0.34, P<0.05; Supplementary Figure S2A), 30–45 min post awakening (r=−0.39, P<0.05; Figure 3a) and 45–60 min post awakening (r=−0.37, P<0.05 Supplementary Figure S2B). There was no correlation between hGRtotal or hGR 1C relative expression and cortisol levels.

(a) Correlation between hGR 1B mRNA relative expression and salivary cortisol levels for the time interval between 30–45 min after awakening. (b) Correlations between total DNA methylation level in GR1B promoter and hGR 1B (gray dots) and hGRtotal mRNA relative expression (black dots). (c) Correlations between hGR 1B mRNA relative expression and DNA methylation level in hGR1B promoter at CpG sites11 (gray dots) and 13 (black dots). (d) Correlation between cortisol levels for the time interval between 30–45 min after awakening and hGR1B promoter DNA methylation levels at CpG sites 11 (gray dots) and 13 (black dots). hGR, human glucocorticoid receptor.
We next assessed whether hGR non-coding exons’ expression differs in current and past PTSD individuals. Our analyses revealed no significant difference in the expression of hGRtotal (F(1,26)=2.277, P=0.145), 1B (F(1,26)=1.611, P=0.217), 1C (F(1,25)=0.102, P=0.752), and 1H (F(1,26)=1.171, P=0.290) between current and past PTSD (Supplementary Figure S1B). These effects were also independent of age and sex.
T-lymphocyte methylation levels
We sought to determine whether alterations in DNA methylation patterns within hGR variants’ promoter could explain the expression changes observed between the PTSD group and controls. We focused on the two variants (hGR 1B and 1C) that showed higher expression in the PTSD group compared with controls (Supplementary Figure S3).
Human glucocorticoid receptor 1B
Methylation levels were analyzed across a region of 479 bp composed of 29 CpGs located upstream hGR 1B (Supplementary Figure S3). Mixed two-way ANOVA revealed a significant main effect of group on percentage of methylation (F(1,182)=28.7, P<0.0001), CpG site (F(15,96)=307.8, P<0.0001), and a group by CpG site interaction (F(15,96)=2.8, P<0.005). Our analysis indicated a significant effect of age (F(1,52)=9.5, P<0.005) on methylation levels although the main effects of group, CpG site, and group by CpG site interaction remained significant after correction for age. In accordance with the expression data, overall methylation level in hGR 1B promoter was significantly lower in PTSD compared with control (Figure 4a). Post hoc tests showed site-specific hypomethylation at CpG sites 2–4 (P<0.005), 11 (P<0.05), 13 (P<0.01) and 14 (P=0.05) in individuals suffering from lifetime PTSD compared with controls (Supplementary Figure S4A).

Overall percentage of methylation (mean±s.e.m.) in (a) hGR 1B, and (b) hGR 1C promoters as a function of group status. PTSD: n=30, Controls: n=16. *P<0.05. CTRL, control; hGR, human glucocorticoid receptor; PTSD, posttraumatic stress disorder.
Our results show a negative correlation between overall methylation levels in the hGR 1B promoter and hGRtotal (r=−0.35, P<0.05) and 1B (r=−0.51, P<0.001) relative expression (Figure 3b). GRtotal and 1B relative expression was also negatively correlated with methylation levels at CpG sites 2–4 (hGRtotal: r=−0.37, P<0.05; hGR 1B: r=−0.55, P<0.005; Supplementary Figure S2C), 11 (hGR 1B: r=−0.34, P<0.05), and 13 (hGR 1B: r=−0.32, P<0.05; Figure 3c). In addition, methylation levels at CpG sites 11 and 13 were positively correlated with cortisol levels obtained between awakening and 30 min post awakening (CpG site 11: r=0.4, P=0.05; CpG site 13: r=0.44, P<0.01; Supplementary Figure S2D), and +30 min and +45 min (CpG site 11: r=0.4, P<0.05; CpG site 13: r=0.43, P<0.01; Figure 3d).
We assessed DNA methylation levels in current and past PTSD samples and found no significant group difference (Supplementary Figure S1C). These effects were also independent of age and sex.
Human glucocorticoid receptor 1C
Methylation levels were analyzed across a 364 bp region composed of 54 CpG upstream hGR 1C (Supplementary Figure S3). Mixed two-way ANOVA revealed a significant main effect of group on percentage of methylation (F(1,354)=6.5, P<0.05), CpG site (F(21,698)=245.6, P<0.0001), but no group by CpG site interaction. In accordance with the expression data, total methylation level in hGR 1C promoter was significantly lower in PTSD compared with the controls (Figure 4b). Post hoc analysis showed significant hypomethylation in PTSD patients at CpG sites 40–41 (P<0.05). We also found one CpG site (CpG 51) to be significantly hypermethylated (P<0.05; Supplementary Figure S4B). An ANCOVA revealed no effect of age and sex on hGR 1C methylation levels between groups.
Our analyses showed no correlation between total methylation in 1C promoter and relative hGR 1C expression or cortisol levels.
We also compared DNA methylation levels in current and past PTSD samples. Our analysis revealed no difference in DNA methylation levels between current and past PTSD (Supplementary Figure S1D). These effects were also independent of age and sex.
Discussion
In this study, we investigated the HPA axis profile of individuals suffering from lifetime PTSD and healthy controls by measuring basal salivary cortisol levels, hGR peripheral expression and CpG methylation states in T lymphocytes. We focused our investigation on the non-coding exons 1B, 1C, 1F and 1H, which have been previously studied in the brain of depressed subjects40 and abused suicide completers.18,19 By combining these three molecular measures, we sought to obtain a comprehensive view of the HPA axis profile and to investigate whether epigenetic mechanisms, in response to traumatic events, modify GR expression and cortisol release in individuals with a diagnosis of lifetime PTSD.
In agreement with the previous findings,13,15,41,42 our results suggest a dysregulation of the HPA axis activity in individuals with PTSD that was independent of current versus past PTSD status. Accordingly, the rise in morning cortisol release, up to 60 min after awakening observed in the control group was lower in the PTSD group. Importantly, the measure of cortisol in this study was obtained from saliva, which has been highly correlated with serum unbound cortisol levels.43 Under normal conditions, unbound cortisol increases by about 50% within the first 30 min after awakening.44 Once released in the blood, 90–95% of cortisol binds corticosteroid binding globulin proteins whereas the remaining fraction enters the cells and activate both the glucocorticoid and mineralocorticoid receptors.45,46 The lower levels of morning salivary cortisol found in this study may indicate that the HPA axis in individuals with lifetime PTSD may be hypoactive. One may assume that this hypoactivity is the consequence of GR hypersensitivity.47
Accordingly, our data showed that hGR expression levels are increased in lifetime PTSD independently of current PTSD status. Although similar results have previously been reported,48 it is the first time that individual hGR variants are investigated in PTSD, and our results suggest that the higher hGR expression may be attributed to the cumulative elevations in hGR 1B and 1C expression. On the other hand, hGR 1H expression was stable between groups and hGR 1F could not be detected. This is in agreement with previous findings showing that hGR variants 1B, 1C and 1H are highly expressed in peripheral blood whereas hGR 1F is expressed almost exclusively in the brain,23 and suggests that modifications in peripheral hGR 1B and 1C expression may be associated with the pathophysiology of PTSD. It is still unclear whether these changes in hGR expression reported in the blood might also be found in the brain. For instance, similar transcriptional differences in the hippocampus (HPC) may alter the inhibitory feedback exerted on the HPA axis activity that would consequently associate with the hypoactivity of the axis reported in this study. More work is required to define whether changes occurring in the peripheral tissue may also be found in the brain.
The expression of specific hGR non-coding exon 1 variants has been associated with different GR protein isoforms.49 These proteins include GRα, GRβ and GR-P. GRα acts as the main functionally active isoform while GRβ and GR-P inhibits and enhances GRα and β activity.50, 51, 52, 53, 54 Interestingly, GRα and GR-P have been associated with hGR 1C and 1B expression, respectively.49 Thus, our results suggest that both GRα and GR-P may be up-regulated in individuals with lifetime PTSD providing a potential explanation for the hypoactivity of the HPA axis found in these patients. In addition, we found a correlation between hGR 1B expression and cortisol levels at awakening, 30 and 45 min after awakening. This suggests that hGR 1B variant may be involved more specifically in the morning increase in cortisol release. Indeed, one may assume that, under normal conditions, the enhancing effect of GR-P may be attenuated to allow more cortisol to be released. However, the overexpression of hGR 1B in PTSD, leading to higher levels of GR-P, may counter this attenuation by potentiating the GR-induced inhibition of the HPA-axis activity and subsequently decreasing morning cortisol release. More work is required to clarify the roles of GR protein isoforms on cortisol release.
Our findings suggest that hGR expression may be regulated by changes in DNA methylation. DNA methylation is an epigenetic mark associated with transcriptional repression that acts by interfering with the binding of transcription factors and with the recruitment of the transcriptional machinery to the DNA.16 Our results show a significant overall and site-specific hypomethylation in the promoters of hGR 1B and 1C in the PTSD group compared to controls. In addition, hGR total and hGR 1B expression was inversely correlated with overall and site-specific methylation levels in hGR 1B promoter. This suggest that lower levels of methylation in hGR 1B promoter may allow the transcriptional machinery to bind within this region and potentiate expression as it has been suggested for multiple hGR variants’ promoters.55
Previous studies suggested that epigenetic mechanisms may be involved in the pathophysiology of PTSD. For instance, DNA methylation alterations in the promoter of the brain-derived neurotrophic factor gene associated with changes in gene expression, have been found within the HPC in a rat model of PTSD.56 In humans, Uddin et al.57 used a genome-wide approach and reported associations between PTSD and methylation patterns in peripheral blood noting that differentially methylated CpG sites were overrepresented in genes related to immune function and inflammation. This was associated with overall DNA methylation levels being inversely correlated with gene expression levels. Collectively, these results suggest that traumatic events may induce alterations in DNA methylation patterns, which may change gene expression patterns and subsequently induce behavioral modifications in PTSD. It should be mentioned that we did not measure GR protein levels in this study and, although mRNA levels are consistently correlated with DNA methylation levels, future studies should consider measuring protein levels to strengthen the functional impact of DNA methylation on biological functions.
Early-life stress has been shown to interfere with gene expression by altering DNA methylation patterns at multiple genes including GR.17, 18, 19,56,58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68 For instance, decreased GR 17 expression associated with promoter hypermethylation has been reported in the HPC of low licking and grooming mothers’ offspring.17 Similarly, lower hGR 1B, 1C and 1F expression has been associated with promoter hypermethylation in the HPC of abused suicide completers18,19 while hGR 1B, 1C, 1D, 1F, 1H and 1J expression patterns have been shown to vary in the brain of depressed individuals with no differences in promoter methylation levels.40 However, our results suggest that trauma exposure together with a lifetime diagnosis of PTSD is associated with hypomethylation of hGR 1B and 1C promoter. Although contrasting, these results are in agreement with the variations in the activity of the HPA axis found in victims of child abuse and depressed individuals compared with lifetime PTSDs. Indeed, the HPA axis in victims of child abuse, as well as depressed patients, is hyperactive.69, 70, 71 In contrast, our results, as well as previous findings,13,15,41,42 show that the HPA axis in PTSD patients is hypoactive and matches with the overexpression of hGR 1B and 1C associated with promoter hypomethylation found in PTSDs. Consequently, this suggests that the consequences of stress on the epigenetic regulation of hGR may differ according to type and timing of stressful events and psychiatric condition. Unfortunately, the design of our study did not allow to adequately control for depression. As such, it is unclear whether or not depressive-like symptoms may partly account for our results. More work will be needed to shed light on the important contribution that depression has on the molecular characterization of PTSD symptoms.
The regulation of the HPA axis takes place mainly in the HPC via the binding of GR by glucocorticoids inducing an inhibitory feedback on the activity of the axis.9,71 However, given that the participants for this study are alive, our experiments were performed in peripheral blood samples, which have been frequently used in studies of PTSD.14,48,57 Individual differences in lymphocyte GR responsiveness have been associated with changes in the HPA-axis activity72 and with PTSD symptoms.73 In addition, T lymphocytes have a major role in cell-mediated immunity and it is believed that traumatic events may induce long lasting epigenetic marks conferring this cell type a memory of past adverse life events. Consequently, we believe that these cells may represent a proxy to the molecular alterations that occur in the brain. More work is required to determine whether similar alterations can be observed in the blood and the brain.
It is important to mention that our analyses revealed no change between current and lifetime PTSD. This may be owing to the small sample size of this exploratory study. Alternately, it is possible that these epigenetic changes, which may be induced by traumatic events, are stable in time and may not be affected by treatment. These changes may also increase vulnerability to stress in individuals with a past PTSD. Epigenetic mechanisms and gene expression in the brain and in peripheral tissue can be affected by antidepressant treatment.65,66,74 However, our results suggest that this may not be the case for the regulation of GR in PTSD. This is also consistent with our previous results obtained from the brain tissue of abused suicide completers.19 Thus, although we did not correct for medication in this study, our analysis comparing current and past PTSD suggest that medication may not have an effect on cortisol levels, GR expression and DNA methylation. Nevertheless, more work is required to study the effect of treatment on epigenetic mechanisms and on the epigenetic control of GR expression in the blood.
To summarize, our results suggest that the expression of hGR 1B and 1C is increased in peripheral blood samples from individuals with lifetime PTSD. These differences in expression were associated with overall and site-specific DNA hypomethylation in T lymphocytes and with lower release of morning salivary cortisol in PTSD. Thus, our results suggest that traumatic events in PTSD induce DNA methylation alterations that modify gene expression and the activity of the HPA axis. However, these alterations differ from the ones described in depressed and abused suicide completers suggesting that different epigenetic mechanisms are involved in the regulation of GR in PTSD individuals. More work is required to better understand the epigenetic mechanisms regulating the expression of GR and the activity of the HPA axis in PTSD.
References
Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB . Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry 1995; 52: 1048–1060.
Breslau N, Kessler RC, Chilcoat HD, Schultz LR, Davis GC, Andreski P . Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. Arch Gen Psychiatry 1998; 55: 626–632.
Van Ameringen M, Mancini C, Patterson B, Boyle MH . Post-traumatic stress disorder in Canada. CNS Neurosci Ther. 2008. Fall 14: 171–181.
Stein MB, Walker JR, Hazen AL, Forde DR . Full and partial posttraumatic stress disorder: findings from a community survey. Am J Psychiatry 1997; 154: 1114–1119.
Resnick HS, Kilpatrick DG, Dansky BS, Saunders BE, Best CL . Prevalence of civilian trauma and posttraumatic stress disorder in a representative national sample of women. J Consult Clin Psychol 1993; 61: 984–991.
Frans O, Rimmo PA, Aberg L, Fredrikson M . Trauma exposure and post-traumatic stress disorder in the general population. Acta Psychiatr Scand 2005; 111: 291–299.
Breslau N . The epidemiology of posttraumatic stress disorder: what is the extent of the problem? J Clin Psychiatry 2001; 62: 16–22.
Mehta D, Binder EB . Gene x environment vulnerability factors for PTSD: The HPA-axis. Neuropharmacology 2012; 62: 654–662.
Heim C, Newport DJ, Mletzko T, Miller AH, Nemeroff CB . The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology 2008; 33: 693–710.
Meaney MJ, Szyf M . Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci 2005; 28: 456–463.
Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK et al. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 1999; 156: 585–588.
Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM et al. Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry 1997; 154: 624–629.
Meewisse ML, Reitsma JB, de Vries GJ, Gersons BP, Olff M . Cortisol and post-traumatic stress disorder in adults: systematic review and meta-analysis. Br J Psychiatry 2007; 191: 387–392.
Yehuda R, Lowy MT, Southwick SM, Shaffer D, Giller EL Jr . Lymphocyte glucocorticoid receptor number in posttraumatic stress disorder. Am J Psychiatry 1991; 148: 499–504.
de Kloet CS, Vermetten E, Geuze E, Kavelaars A, Heijnen CJ, Westenberg HG . Assessment of HPA-axis function in posttraumatic stress disorder: pharmacological and non-pharmacological challenge tests, a review. J Psychiatr Res 2006; 40: 550–567.
Klose RJ, Bird AP . Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 2006; 31: 89–97.
Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004; 7: 847–854.
McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 2009; 12: 342–348.
Labonte B, Yerko V, Gross J, Mechawar N, Meaney MJ, Szyf M et al. Differential glucocorticoid receptor exon 1(B), 1(C), and 1(H) expression and methylation in suicide completers with a history of childhood abuse. Biol Psychiatry 2012; 72: 41–48.
Zong J, Ashraf J, Thompson EB . The promoter and first, untranslated exon of the human glucocorticoid receptor gene are GC rich but lack consensus glucocorticoid receptor element sites. Mol Cell Biol 1990; 10: 5580–5585.
Encio IJ, Detera-Wadleigh SD . The genomic structure of the human glucocorticoid receptor. J Biol Chem 1991; 266: 7182–7188.
Breslin MB, Geng CD, Vedeckis WV . Multiple promoters exist in the human GR gene, one of which is activated by glucocorticoids. Mol Endocrinol 2001; 15: 1381–1395.
Turner JD, Muller CP . Structure of the glucocorticoid receptor (NR3C1) gene 5' untranslated region: identification, and tissue distribution of multiple new human exon 1. J Mol Endocrinol 2005; 35: 283–292.
Strahle U, Schmidt A, Kelsey G, Stewart AF, Cole TJ, Schmid W et al. At least three promoters direct expression of the mouse glucocorticoid receptor gene. Proc Natl Acad Sci USA 1992; 89: 6731–6735.
Pedersen KB, Geng CD, Vedeckis WV . Three mechanisms are involved in glucocorticoid receptor autoregulation in a human T-lymphoblast cell line. Biochemistry 2004; 43: 10851–10858.
Barrett TJ, Vig E, Vedeckis WV . Coordinate regulation of glucocorticoid receptor and c-jun gene expression is cell type-specific and exhibits differential hormonal sensitivity for down- and up-regulation. Biochemistry 1996; 35: 9746–9753.
Wei P, Vedeckis WV . Regulation of the glucocorticoid receptor gene by the AP-1 transcription factor. Endocrine 1997; 7: 303–310.
Breslin MB, Vedeckis WV . The human glucocorticoid receptor promoter upstream sequences contain binding sites for the ubiquitous transcription factor, Yin Yang 1. J Steroid Biochem Mol Biol 1998; 67: 369–381.
Nunez BS, Vedeckis WV . Characterization of promoter 1B in the human glucocorticoid receptor gene. Mol Cell Endocrinol 2002; 189: 191–199.
Geng CD, Vedeckis WV . Steroid-responsive sequences in the human glucocorticoid receptor gene 1A promoter. Mol Endocrinol 2004; 18: 912–924.
Labonte B, Turecki G . The epigenetics of suicide: explaining the biological effects of early life environmental adversity. Arch Suicide Res 2010; 14: 291–310.
Deaton AM, Webb S, Kerr AR, Illingworth RS, Guy J, Andrews R et al. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res 2011; 21: 1074–1086.
United States Department of Veterans Affairs Employee Education System, National Center for Post-Traumatic Stress Disorder (U.S.). The Clinician Administered PTSD Scale (CAPS) for DSM-IV an Independent Study. Department of Veterans Affairs, Employee Education System: St Louis, MO, USA, 2001.
Brunet A, St-Hilaire A, Jehel L, King S . Validation of a French version of the impact of event scale-revised. Can J Psychiatry 2003; 48: 56–61.
Derogatis LR Savitz KL The SCL-90-R and the Brief Symptom Inventory (BSI) in Primary Care. Maruish ME (eds). Handbook of Psychological Assessment in Primary Care Settings. Lawrence Erlbaum Associates: Mahwah, NJ, USA, 2000, 297–334.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 1998; 59 Suppl 20: 22–33. quiz 34–57.
Hellhammer DH Kirschbaum C Belkien L Measurement of salivary cortisol under psychological stimulation. Hingtgen N, Hellhammer DH, Huppmann G (eds). Advanced Methods in Psychobiology. Hogrefe: Toronto, ON, Canada, 1987, 280–289.
Dressendorfer RA, Kirschbaum C, Rohde W, Stahl F, Strasburger CJ . Synthesis of a cortisol-biotin conjugate and evaluation as a tracer in an immunoassay for salivary cortisol measurement. J Steroid Biochem Mol Biol 1992; 43: 683–692.
Pruessner JC, Kirschbaum C, Meinlschmid G, Hellhammer DH . Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology 2003; 28: 916–931.
Alt SR, Turner JD, Klok MD, Meijer OC, Lakke EA, Derijk RH et al. Differential expression of glucocorticoid receptor transcripts in major depressive disorder is not epigenetically programmed. Psychoneuroendocrinology 2010; 35: 544–556.
Neylan TC, Brunet A, Pole N, Best SR, Metzler TJ, Yehuda R et al. PTSD symptoms predict waking salivary cortisol levels in police officers. Psychoneuroendocrinology 2005; 30: 373–381.
Wessa M, Rohleder N, Kirschbaum C, Flor H . Altered cortisol awakening response in posttraumatic stress disorder. Psychoneuroendocrinology 2006; 31: 209–215.
Umeda T, Hiramatsu R, Iwaoka T, Shimada T, Miura F, Sato T . Use of saliva for monitoring unbound free cortisol levels in serum. Clin Chim Acta 1981; 110: 245–253.
Pruessner JC, Wolf OT, Hellhammer DH, Buske-Kirschbaum A, von Auer K, Jobst S et al. Free cortisol levels after awakening: a reliable biological marker for the assessment of adrenocortical activity. Life Sci 1997; 61: 2539–2549.
Siiteri PK, Murai JT, Hammond GL, Nisker JA, Raymoure WJ, Kuhn RW . The serum transport of steroid hormones. Recent Prog Horm Res 1982; 38: 457–510.
Kumsta R, Entringer S, Hellhammer DH, Wust S . Cortisol and ACTH responses to psychosocial stress are modulated by corticosteroid binding globulin levels. Psychoneuroendocrinology 2007; 32: 1153–1157.
Pruessner JC, Gaab J, Hellhammer DH, Lintz D, Schommer N, Kirschbaum C . Increasing correlations between personality traits and cortisol stress responses obtained by data aggregation. Psychoneuroendocrinology 1997; 22: 615–625.
Yehuda R, Boisoneau D, Lowy MT, Giller EL Jr . Dose-response changes in plasma cortisol and lymphocyte glucocorticoid receptors following dexamethasone administration in combat veterans with and without posttraumatic stress disorder. Arch Gen Psychiatry 1995; 52: 583–593.
Russcher H, Dalm VA, de Jong FH, Brinkmann AO, Hofland LJ, Lamberts SW et al. Associations between promoter usage and alternative splicing of the glucocorticoid receptor gene. J Mol Endocrinol 2007; 38: 91–98.
Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985-1986; 318: 635–641.
Bamberger CM, Bamberger AM, de Castro M, Chrousos GP . Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest 1995; 95: 2435–2441.
Oakley RH, Sar M, Cidlowski JA . The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J Biol Chem 1996; 271: 9550–9559.
Krett NL, Pillay S, Moalli PA, Greipp PR, Rosen ST . A variant glucocorticoid receptor messenger RNA is expressed in multiple myeloma patients. Cancer Res 1995; 55: 2727–2729.
de Lange P, Segeren CM, Koper JW, Wiemer E, Sonneveld P, Brinkmann AO et al. Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor-mediated effects in transfected cells. Cancer Res 2001; 61: 3937–3941.
Cao-Lei L, Leija SC, Kumsta R, Wust S, Meyer J, Turner JD et al. Transcriptional control of the human glucocorticoid receptor: identification and analysis of alternative promoter regions. Hum Genet 2011; 129: 533–543.
Roth TL, Zoladz PR, Sweatt JD, Diamond DM . Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J Psychiatr Res 2011; 45: 919–926.
Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R et al. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci USA 2010; 107: 9470–9475.
Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ . Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology 2006; 147: 2909–2915.
Ernst C, Chen ES, Turecki G . Histone methylation and decreased expression of TrkB.T1 in orbital frontal cortex of suicide completers. Mol Psychiatry 2009; 14: 830–832.
McGowan PO, Sasaki A, Huang TC, Unterberger A, Suderman M, Ernst C et al. Promoter-wide hypermethylation of the ribosomal RNA gene promoter in the suicide brain. PLoS One 2008; 3: e2085.
McGowan PO, Suderman M, Sasaki A, Huang TC, Hallett M, Meaney MJ et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One 2011; 6: e14739.
Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmuhl Y, Fischer D et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci 2009; 12: 1559–1566.
Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A et al. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl Psychiatry 2011; 1: 1–6.
Roth TL, Lubin FD, Funk AJ, Sweatt JD . Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry 2009; 65: 760–769.
Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ . Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci 2006; 9: 519–525.
Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T et al. Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 2011; 69: 359–372.
Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ . Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci 2010; 30: 13130–13137.
Labonte B, Suderman M, Maussion G, Navaro L, Yerko V, Mahar I et al. Genome-wide epigenetic regulation by early-life trauma. Arch Gen Psychiatry 2012; 69: 722–731.
Heim C, Mletzko T, Purselle D, Musselman DL, Nemeroff CB . The dexamethasone/corticotropin-releasing factor test in men with major depression: role of childhood trauma. Biol Psychiatry 2008; 63: 398–405.
Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA 2000; 284: 592–597.
Nemeroff CB, Vale WW . The neurobiology of depression: inroads to treatment and new drug discovery. J Clin Psychiatry 2005; 66: 5–13.
Yehuda R, Yang RK, Guo SL, Makotkine I, Singh B . Relationship between dexamethasone-inhibited lysozyme activity in peripheral mononuclear leukocytes and the cortisol and glucocorticoid receptor response to dexamethasone. J Psychiatr Res 2003; 37: 471–477.
Yehuda R, Engel SM, Brand SR, Seckl J, Marcus SM, Berkowitz GS . Transgenerational effects of posttraumatic stress disorder in babies of mothers exposed to the World Trade Center attacks during pregnancy. J Clin Endocrinol Metab 2005; 90: 4115–4118.
Lopez JP, Mamdani F, Labonte B, Beaulieu MM, Yang JP, Berlim MT et al. Epigenetic regulation of BDNF expression according to antidepressant response. Mol Psychiatry 2012; 18: 398–399.
Acknowledgements
GT and AB obtained a salary award from the Fonds de Recherche du Québec en santé (FRQ-S). BL is supported by a Frederick Banting and Charles Best doctoral fellowship from the Canadian Institutes of Health Research (CIHR).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Labonté, B., Azoulay, N., Yerko, V. et al. Epigenetic modulation of glucocorticoid receptors in posttraumatic stress disorder. Transl Psychiatry 4, e368 (2014). https://doi.org/10.1038/tp.2014.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.3
Keywords
This article is cited by
-
Epigenetics of traumatic stress: The association of NR3C1 methylation and posttraumatic stress disorder symptom changes in response to narrative exposure therapy
Translational Psychiatry (2023)
-
Decreased mononuclear cell NR3C1 SKA2 and FKPB5 expression levels among adult survivors of suicide bombing terror attacks in childhood are associated with the development of PTSD
Molecular Psychiatry (2023)
-
Methylation and expression of glucocorticoid receptor exon-1 variants and FKBP5 in teenage suicide-completers
Translational Psychiatry (2023)
-
Aberrant Histone Modification of TNFAIP3, TLR4, TNIP2, miR-146a, and miR-155 in Major Depressive Disorder
Molecular Neurobiology (2023)
-
Evaluation of cross-platform compatibility of a DNA methylation-based glucocorticoid response biomarker
Clinical Epigenetics (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}