
Abstract
Bipolar disorder (BP) is a chronic psychiatric condition characterized by dynamic, pathological mood fluctuations from mania to depression. To date, a major challenge in studying human neuropsychiatric conditions such as BP has been limited access to viable central nervous system tissue to examine disease progression. Patient-derived induced pluripotent stem cells (iPSCs) now offer an opportunity to analyze the full compliment of neural tissues and the prospect of identifying novel disease mechanisms. We have examined changes in gene expression as iPSC derived from well-characterized patients differentiate into neurons; there was little difference in the transcriptome of iPSC, but BP neurons were significantly different than controls in their transcriptional profile. Expression of transcripts for membrane bound receptors and ion channels was significantly increased in BP-derived neurons compared with controls, and we found that lithium pretreatment of BP neurons significantly altered their calcium transient and wave amplitude. The expression of transcription factors involved in the specification of telencephalic neuronal identity was also altered. Control neurons expressed transcripts that confer dorsal telencephalic fate, whereas BP neurons expressed genes involved in the differentiation of ventral (medial ganglionic eminence) regions. Cells were responsive to dorsal/ventral patterning cues, as addition of the Hedgehog (ventral) pathway activator purmorphamine or a dorsalizing agent (lithium) stimulated expression of NKX2-1 (ventral identity) or EMX2 (dorsal) in both groups. Cell-based models should have a significant impact on our understanding of the genesis and therefore treatment of BP; the iPSC cell lines themselves provide an important resource for comparison with other neurodevelopmental disorders.
Similar content being viewed by others


Introduction
Bipolar disorder (BP) is a severe neuropsychiatric disease characterized by recurrent episodes of manias, pathologically energized states with misguided volition and behavior in a mood state of intoxicating euphoria (or irritability) and depressions that are low moods with compromised energy, volitional states and diminished cognitive capacity. The social, personal and vocational consequences are frequently disastrous,1 and the WHO ranks BP among the top causes of lifelong disability.2
The heritability of BP is estimated at 80% and adoption, twin and family studies have consistently identified a significant genetic contribution;3, 4 many genes5 and genetic mechanisms6 have been implicated in BP, consistent with multiple disease loci, each contributing modest effects.7,8 Genome-wide association studies have identified a few replicated loci including polymorphisms in CACNA1C, the alpha subunit of the L-type voltage-gated calcium channel, the strongest and most replicated risk.5,9, 10, 11, 12, 13 Additional replicated polymorphisms have been identified in ANK3, which regulates assembly of voltage-gated sodium channels at the node of Ranvier,14,15 and SYNE1, which has a critical role in synaptic clustering.16 Overall, available data suggest that alterations in membrane organization and/or ion channelopathies may contribute to BP; in fact, dysregulation of Ca2+ signaling is one of the most reproducible observations in BP.17
Like many complex central nervous system (CNS) disorders, the roles of neuropathology, environment and trauma in BP are largely unknown; there are reports of ventricular enlargement, gray matter, hippocampal and cerebellar hypoplasia, and anomalies of white matter tracts,18, 19, 20, 21 that are nonspecific and inconsistent. There is also evidence of abnormal neuronal cell migration22 and of altered adult neurogenesis.23,24 Because viable brain tissue is unavailable, and there is no well-defined genetic event or neuropathology, there are simply no good neural models to study BP.
Since subtle alterations in developmental pathways can produce neurological consequences that only become apparent much later in life,25,26 a developmental model to study neuronal differentiation in BP is required. Because they have tri-lineage differentiation potential similar to human embryonic stem cells (hESCs), induced pluripotent stem cells (iPSCs) provide the opportunity to study the stepwise differentiation of patient-derived cells into neurons and glia to identify alterations in cell behavior and test novel therapeutic approaches.27,28 In BP, they offer the unique opportunity to obtain a robust source of cells from individuals with prospective longitudinal observations, individuals on or off medications, patients who have had multiple episodes, different diagnoses and treatments, unlike hESC. iPSCs have been derived from patients with neurodegenerative disorders29, 30, 31, 32, 33, 34 and other neuropsychiatric disorders,35, 36, 37, 38 but are just being developed for BP.
The current investigation demonstrates that iPSC from BP patients are similar to controls (C) in their tri-lineage differentiation capacity and gene expression profiles; however, the expression of membrane receptors and ion channel transcripts was significantly increased in BP neurons, and different sets of neuronal lineage-specific transcription factors were induced. Consistent with changes in membrane composition, exposure of BP neurons to lithium produced significant alterations in wave amplitude and calcium transient compared with control neurons. Transcripts involved in dorsal telencephalic fate (the default identity) were enriched in control neurons, whereas there was an increase in genes involved in ventral fate and GABAergic interneuron differentiation in BP neurons. The dorsal/ventral neuronal identity of both groups could be influenced by exposure of iPSC to dorsalizing or ventralizing factors. This work provides the first information on gene expression during neuronal differentiation in BP, a rich resource to identify alterations in the differentiation of glia and neurons, as well as an opportunity for comparisons with other neuropsychiatric disorders.
Materials and methods
Patient population
The clinical sample was obtained from the Prechter Longitudinal Study of Bipolar Disorder.39 Subjects contributing a skin sample were from a psychiatric clinic in a mid-western college city, Caucasian, and were diagnosed with Bipolar 1 Disorder, or healthy unaffected controls were ascertained through advertising on the University of Michigan Clinical Studies website. All participants were assessed clinically using the Diagnostic Interview for Genetic Studies.40 BP patients were determined by clinical evaluation to have a chronic disorder with mood symptoms most of the time, age of onset ranging from 15 to 23 years. Two of three Bipolar 1 Disorder patients were considered lithium responsive and all had exposures to antidepressant, anticonvulsants and antipsychotic medications. One patient had comorbid panic disorder and a second patient was treated for metabolic syndrome with a statin (rosuvastatin) and an oral hypoglycemic (pioglitazone). A second patient was receiving levothyroxine. Two patients and two controls were 50±5 years age and the other pair were aged 30 and 34 years at time of sample. Four patients were male and two were female.
Fibroblast derivation
Skin biopsies were obtained in the UM Clinical Research Unit. Samples were dissociated in trypsin, expanded in fibroblast medium comprising Dulbecco’s Modified Eagle Medium (DMEM, 11965–092; Invitrogen, Grand Island, NY, USA) containing 10% heat-inactivated fetal bovine serum (Invitrogen, 10082–147), 1% non-essential amino acids (NEAA) (Invitrogen, 11140–050), 2 mM glutamax (Invitrogen, 35050–061) and 1 × Penicillin/Streptomycin solution (Invitrogen, 15140–122) to obtain dermal fibroblasts. Stock fibroblasts from each patient were banked and cells were analyzed for their expression of fibroblast-restricted genes, pluripotency factors, lineage markers and mycoplasma.
Virus preparation
Retrovirus was made by transfecting 293FT cells with pUMVC and pCMV-VSV-g plasmids, and individual plasmids: pMXs-hKLF4, pMXs-hSOX2, pMXs-hOCT4 or pMXs-hcMYC (Addgene, Cambridge, MA, USA) at a 1:1:1 ratio using Lipofectamine 2000 (Invitrogen, 11668-019). Virus supernatant was collected and combined, filtered through a 0.45-μm filter (EMD Millipore, SCHVU01RE, Billerica, MA, USA) and concentrated 80–100-fold in a centrifugal filter with a 1 00 000 MWCO (EMD Millipore, UFC910008). The concentrate containing all four factors was then used immediately to transduce fibroblasts at a final concentration between 3- and 12-fold.
iPSC derivation
Fibroblasts at passage 5–6 were plated at 5 × 104 cells in 35-mm tissue culture dishes in fibroblast medium and transduced with retroviral constructs expressing the pluripotency factors OCT4, SOX2, KLF4 and c-MYC.41 Fibroblasts were transduced on day 1 after plating, with daily medium changes from day 3 to day 6. On day 7, forming iPSC were trypsinized and passaged to mouse embryonic fibroblast (MEF) feeders for expansion in hESC medium compromising DMEM/F12 (Invitrogen, 11320–032), 20% KOSR (Invitrogen, 10828–028), 4 ng ml−1 FGF2 (EMD Millipore GF003), 1% NEAA, 2 mM glutamax and 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St Louis, MO, USA, M3148). After 10 days on MEFs, medium was changed to MEF-conditioned hESC medium, then hESC-like colonies manually dissected and passaged to new MEFs for expansion in hESC medium. From each fibroblast line, 5–10 colonies were selected for expansion to iPSC lines; a minimum of 5–6 iPSC lines were derived from each fibroblast (patient) sample.
Pluripotency characterization
Analysis of the expression of pluripotency factors NANOG, SOX2 and OCT4, and cell surface expression of SSEA3, SSEA4, TRA-1–60 and TRA-1–81, and alkaline phosphatase was carried out in immunocytochemistry and polymerase chain reaction (PCR) (Supplementary Figure 1). Controls for pluripotency and differentiation capability include parallel analysis of a well-characterized iPSC line (UM-C2) and H7 or UM4-6 hESC lines.
Differentiation capacity
Assessment of the tri-lineage differentiation capability of the iPSC lines was done by 45 days of culture in non-adherent dishes in hESC medium lacking FGF2 to form embryoid bodies (EBs). Lineage differentiation was assessed using cell-type restricted antibodies and PCR (Supplementary Figure 1D,Supplementary Tables 1A and B). Ectoderm was identified using nestin or β-III tubulin; mesoderm: Brachyury, smooth muscle actin or decorin; and endoderm: Sox17, alpha-fetoprotein or Foxa2.
Neuronal differentiation
iPSCs were manually dissected from MEF, then grown in suspension culture for 4 days in EB medium supplemented with 2 μM dorsomorphin (R&D Systems, 3096; Minneapolis, MN, USA) and 5 μM SB431542 (R&D Systems, 1614). Medium was then changed to neural induction medium consisting of DMEM/F12, 1 × N2 supplement (Invitrogen, 17502-048), 1 × NEAA, 2 mM glutamax, 0.1 mM β-mercaptoethanol, 2 μM dorsomorphin, 5 μM SB431542 and 20 ng ml−1 FGF2 for an additional 3 days in suspension. Neurospheres were then planted on laminin (Sigma-Aldrich, L2020)-coated culture dishes for rosette formation. Rosettes were manually picked 2–3 times, dissociated in Accutase then re-plated on poly-D-ornithine-laminin-coated substrates for neuronal differentiation in medium composed of neural basal medium (Invitrogen, 21103–049), 1 × B27 supplement (Invitrogen, 17504–044), 1 mM glutamine, 1% NEAA and 1 × Penicillin/Streptomycin for an additional 4, 8 or 12 weeks. Medium was changed every other day. To determine whether dorsal–ventral fate could be respecified, cells were grown with the Smoothened agonist (Hedgehog pathway activator) purmorphamine (1 μm; Cayman Chemical, Ann Arbor, MI, USA, 10009634), or with the dorsalizing agent lithium chloride (LiCl; 1 mM; Sigma-Aldrich). Controls were exposed to DMSO (carrier) alone.42,43
Reverse transcriptase-polymerase chain reaction
Total RNA from iPSCs, EBs or neurons was extracted with Trizol (Invitrogen, 15596-0926), ±5 μg of UltraPure glycogen (Thermo Fisher Scientific, Waltham, MA, USA) to enhance RNA recovery. RNA was treated with DNase (Thermo Fisher Scientific) and reverse transcribed (Superscript III, Invitrogen) using random nonamers. The cDNA was then used in PCR (primers in Supplementary Table 1B).
Microarray analysis
To characterize the iPSC and to determine whether there were differences in their gene expression profiles with neuronal differentiation, total RNA was isolated from six individual iPSC cell lines (3 BP patients and 3 controls (C)) before and following 8 weeks of neuronal differentiation using the TRIzol reagent (Invitrogen, 15596-026). RNAs were DNased followed by the RNeasy MinElute Cleanup reagent (Qiagen, Hilden, Germany, 74204). RNA quantity and quantity were measured in an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) producing RNAs with an integrity measure > 9.5. They were amplified and hybridized to GeneChip U133 Plus 2.0 microarrays (Affymetrix, Santa Clara, CA, USA). Data quality control parameters included scaling factor, noise, background, percentage of present calls, 5′/3′ signal ratios observed in GAPDH mRNA and 5′/3′ signal ratios of spiking genes. Raw data (U133P2 CEL files) were processed using the Bioconductor package ‘Affy’ for R. Normalized Log2 transformed probe intensity Robust Multi-chip Average values were used in differential gene expression analyses and transcripts with significantly altered expression (P⩽0.05) between BP and controls were identified using t-test with R and David software.44 To assess similarities between groups, the Symmetrized Stein Distance was calculated between estimated sample covariance matrices for each sample pair (BP vs control iPSC, BP vs control neurons and iPSC vs neurons). Statistical significance was calculated using the permutation test.45 Validation of the microarray results was performed with TaqMan ‘Assays on Demand’ (ABI, Foster City, CA, USA) for seven genes selected based on fold changes between BP and control samples, using the 2−ΔΔCt method.46 Only complete sets of iPSC (3 BP and 3 C patients) and neurons (same 3 BP and same 3 C cell lines from 6 individuals) were analyzed to minimize stochastic changes due to culture conditions.
Calcium signaling
To simultaneously measure action potential and wave propagation, we differentiated neurons as described above for an additional 4–8 weeks, loaded them with the intracellular calcium-sensitive dye Fluo-4AM, (10 mM, Molecular Probes, Eugene, OR, USA). After 30 min, non-bound dye was removed and cells were examined in a Nikon A1R laser scanning confocal microscope system (Nikon Corporation, Chiyoda, Tokyo, Japan) to identify spontaneous activity. To evoke action potentials, 50 mM KCl was added to the dish and signaling was recorded.47 To determine whether lithium pretreatment would alter signaling, sets of neurons (3 BP and 3 C) were cultured in the presence of LiCl (1 mM) for 24 h before loading and image acquisition. For image analysis, regions of interest were defined as cell bodies or axons, and wave amplitude and propagation were measured from 5 to 10 regions per sample.
Results
Cells
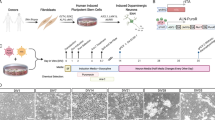
Fibroblast lines have been derived from 22 BP and 10 C patients, expanded and banked to 12 stock vials (or cryostraws) from each patient. From 3 BP and 3 C fibroblast cell lines, we have reprogrammed at least 3 iPSC lines per individual (BP iPSC=28; C iPSC=14). With reprogramming, fibroblast-restricted genes were downregulated (Figures 1a–c; Supplementary Figure 1A) and expression of pluripotency factors was induced (Figure 1d; Supplementary Figures 1B and C). Cells expressing markers of all three germ layers (ectoderm, endoderm and mesoderm) formed following EB differentiation (Supplementary Figure 1D). With differentiation of neurospheres (Figure 1e) followed by neural rosettes (Figure 1f) Sox1, Sox2, Sox3, Pax6 and nestin were expressed (Figure 1g, Supplementary Figure 2). After 4 weeks, neurons with dendrites and axons expressing MAP2, TauAB and β-III tubulin (Figure 1h) formed. Neurons adopted a largely forebrain phenotype, with VGlut+ pyramidal-shaped neurons, GABA+ interneurons, scattered serotonergic and dopaminergic neurons present in the cultures. Many expressed punctate Synapsin1 along their neurites (Figure 1i), with little glial fibrillary acidic protein (GFAP) in the cultures.

Derivation, characterization and neuronal differentiation of iPSC. Fibroblasts (a) express Te-7 (b; Cy3-secondary antibody, red). Reprogrammed colonies of iPSC form and are passaged to MEFs. They downregulate Te-7 (c; Cy3-secondary antibody), and exhibit nuclear expression of Nanog (d; FITC-secondary antibody). For differentiation, cells are removed from MEF and grown in defined medium as neurospheres (e), then plated to form rosettes (f). Rosettes are picked twice, then differentiated in adherent culture in defined medium forming nestin + (g; FITC-secondary antibody) neural progenitor cells, and with additional time in vitro undergo widespread differentiation to β-III tubulin + neurons (h; Cy3-secondary antibody), with punctate expression of Synapsin 1 (Syn1) along their processes (i, Cy3-secondary antibody). All cells, but a, are stained with Hoechst 23487 (blue) to identify nuclei. Scale bars in a–d,g = 500 mm; e = 2 mm, f,h,i = 200 mm.
Microarray analysis
Pooled data: BP and control
Analysis of the 54 676 probesets identified 20 550 unique transcripts expressed in iPSC and 20 718 in neuronally differentiated cells. We replicated five out of seven genes selected from our microarray in quantitative reverse transcriptase-polymerase chain reaction, supporting the validity of these results (Supplementary Table 1C). In the iPSC samples, 2538 transcripts were expressed at significantly different levels (Supplementary Figure 3A). KEGG pathway analysis identified one significant pathway in the iPSC, calcium signaling, n=39, P⩽2.5 × 10−2, Bonferroni correction. Many of these transcripts are associated with neuroplasticity and development, others with stress-activated pathways (Supplementary Table 2). Of the 20 718 transcripts identified in neuronally differentiated iPSC, 1835 transcripts were differentially expressed between groups (nominal P⩽0.05), but KEGG pathway analysis did not identify significantly altered pathways in the neuronal population. Additional analysis of the differentially expressed transcripts did not identify significant differences between BP vs control iPSC (distance=0.036, P⩽0.072), not surprisingly pooled iPSC vs neurons were significantly different (distance=0.174, P⩽0.012). Interestingly, BP and control neurons were also significantly different in their transcriptomes (distance=0.103, P⩽0.041); Supplementary Figure 3B.
Within-groups analysis: BP iPSC vs BP neurons and control iPSC vs control neurons
Transcripts expressed in iPSC and neurons There were 14 993 unique transcripts that were significantly differentially expressed in BP iPSC compared with neurons derived from them; there were 15 311 transcripts in control iPSC vs control neurons that were significantly altered. Of these, 7242 transcripts were expressed at twofold or higher levels in BP iPSC, 5120 were expressed at ⩾ twofold levels in BP-derived neurons. In control cells, 7098 unique transcripts were expressed at twofold or higher levels in iPSC and 5089 at ⩾ twofold levels in neurons.
i. Transcripts enriched in iPSC Functional Annotation Chart Analysis identified 475 Gene Ontology (GO) terms associated with genes expressed at significantly higher levels in BP iPSC than in differentiated BP neurons; 463 terms in control iPSC vs neurons. These included transcripts involved in cell cycle control, RNA binding and genes involved in development (Table 1 summarizes clusters involved in early differentiation).
Both sets of iPSC express similar pluripotency factors, positive regulators of cell cycle, genes involved in very early embryonic development, signaling molecules, and factors involved in proliferation. They express few anterior-posterior patterning factors (HOX) and numerous genes involved in chromatin modification. There were slight variations in the expression of individual transcripts between groups, but the functional significance, if any, of these differences is not known.
ii. Transcripts enriched in neurons Transcripts associated with 425 Gene Ontology categories were significantly increased in BP neurons compared with BP iPSC; 437 in control neurons compared with control iPSC (Table 2 describes categories involved in neuronal differentiation).
With neuronal differentiation, transcripts associated with pluripotency, positive control of the cell cycle, and many chromatin remodeling factors were downregulated, whereas genes involved in signaling, transcripts associated with anterior-posterior patterning (HOX genes) and SOX genes were induced with differentiation. Cell surface molecules, many involved in neurite outgrowth, and transcripts that encode membrane channels were also upregulated with neuronal differentiation. Transcripts involved in early neuronal differentiation including ELAVL1,2,3,4; FOXP1; MAP2; MAPT; MBD2; NEFL, NEFM and NRG1–4 were increased, whereas genes associated with glial lineage differentiation were not identified in BP, but GFAP was present in controls. Transcripts previously suggested to be dysregulated in BP, including ANK3, CACNA1C, GSK3β (glycogen synthase kinase 3 beta), MECP2, NCAN, SYNE1 and ZNF804A, were significantly upregulated as both control and BP iPSC differentiated to neurons, whereas BDNF and DISC1 were not changed.
The ribonuclease DICER1, which is involved in microRNA processing, was significantly higher in BP but not control neurons. Factors involved in ventral neuronal fate such as NKX2-1 and PAX2 were significantly induced in BP, whereas transcripts involved in dorsal fate such as EMX2 (dorsal telencephalon), and HOX genes (HOXA1,6,7) required for positional identity and regional differentiation of the hindbrain were significantly higher in control neurons.
These data demonstrate that with neuronal differentiation, cells withdraw from cell cycle, express different sets of signaling molecules than iPSC and express largely overlapping sets of neuronal lineage markers. Enrichment of dorsal telencephalic transcripts, such as PAX6 and EMX2, and the cortical determinate LHX2 (ref.48) in control and expression of ventral determinates, such as PAX2 and NKX2-1 (basal forebrain progenitors49), involved in formation of the medial ganglionic eminence and GABAergic interneuron differentiation50 in BP neurons suggest that neuronal identity is altered, as mutual inhibition between PAX6 and NKX2-1 determines dorsal/ventral boundaries in the telencephalon.51
Between-groups comparisons: BP vs control cells
Analysis of pluripotent cells There were 2547 unique transcripts expressed at significantly different levels in iPSC derived from BP vs control individuals.
i. Transcripts enriched in BP vs control iPSC One-hundred eighty-six transcripts were expressed at 1.5-fold or greater levels in iPSCs derived from individuals with BP compared with control iPSCs.
There were 14 significant clusters of genes (Table 3i) in this group. Many transcripts previously associated with neurodevelopmental/neuropsychiatric disorders or susceptibility loci for BP were identified (Supplementary Tables 3–7).
ii. Transcripts enriched in control vs BP iPSC Forty-six transcripts were expressed 1.5-fold or higher in iPSC from control vs BP individuals, including genes involved in cell polarity and in lineage differentiation, including the TOR complex2 factor RICTOR, which regulates cell growth. There was one cluster of transforming growth factor-β-related transcripts that was significantly increased (Table 3ii). Members of this cluster included GDF3 and INHBE that have been associated with proliferation and pluripotency and are expressed in the ICM and/or in hESC,52 whereas LEFTY1 and LEFTY2 are involved in nodal inhibition, neural induction and early embryo patterning.53 None was previously associated with psychiatric or human neurodevelopmental disorders. These data indicate that iPSC from BP and control cells are strikingly similar in their transcriptional repertoire. No genes associated with the core regulatory mechanisms that maintain pluripotency were differentially expressed in these two populations.
iii. Genes enriched in BP vs control neurons Of the 1312 unique transcripts that were altered in neurons, 140 transcripts were increased 1.5-fold or greater in BP, including transcripts associated with cell–cell and/or cell–matrix interactions and with axon outgrowth, with neuronal differentiation, organization of synapses, secreted growth factors, and neurotransmitters and their receptors. Many membrane-associated channel genes, transcripts involved in methylation and control of gene expression and with the cytoskeleton were also increased in BP neurons. Functional Annotation Cluster Analysis identified plasma membrane part and ion transport as significantly increased in BP neurons (Table 4iii). Both clusters (Supplementary Tables 8 and 9) contained transcripts previously associated with neurodevelopmental/neuropsychiatric disorders, as well as BP susceptibility loci.
iv. Enriched in control vs BP neurons Ninety-five transcripts were expressed 1.5-fold or higher in control vs BP neurons, including genes involved in cytoskeletal organization and function, cell surface/extracellular matrix factors, apoptosis, DNA repair, in cell cycle, and in signaling. Genes involved in early neuronal differentiation and in dorsal patterning of the CNS including EMX2, EOMES, TCF3 as well as VGLUT1 (SLC17A7; previously reported to be decreased in BP54) were enriched in control but not BP neurons; there was a single significant cluster of nuclear factors (Table 4iv; Supplementary Table 10).
Several patterning factors were altered in BP vs control neurons. HOXA1, A6 and A7 that are expressed in the cerebellum and brainstem were significantly increased in control neurons, consistent with clinical reports of cerebellar hypoplasia20,21,55 in BP. Of transcripts associated with circadian rhythm, TIPIN, PENK and TAC1 were expressed at significantly higher levels in BP vs control neurons; whereas in iPSC NPAS2 was expressed at significantly lower levels in BP vs controls; CRY1 and RORA were expressed at higher levels.
Comparisons with other data sets
We previously examined gene expression in postmortem RNA samples from the premotor cortex of controls, from individuals with BP taking typical antipsychotic medications (BP+), and from unmedicated patients with BP (BP−). Remarkably, gene expression profiles of BP+ were more similar to controls than to BP−.56 When differentially expressed transcripts from BP− brain were compared with those from BP iPSC-derived neurons, 151 significantly altered genes were identified; 57 genes were expressed in a similar direction (χ2=651; P⩽1 × 10−7) including transcripts involved in cell migration, calcium signaling and neurotransmitter receptors (Supplementary Table 11).
Compared with the genes enriched in schizophrenia patient-derived iPSC neurons,35 nine were significantly different in bipolar vs control neurons in our study. SOX6 and FGFR3 were downregulated in both, whereas FGF14, LGALS8, PDE4D, RHOBTB3, SLC41A2, UNC5C and ZMAT4 were expressed at higher levels in schizophrenia and BP than in control neurons. Compared with an analysis of neurons differentiated from iPSC from patients with Timothy Syndrome (CACNA1C mutation37), 233 genes were expressed in a similar pattern as in BP neurons, including many transcripts involved in neuronal differentiation. Clearly, it will be important to compare our analyses with those of additional BP tissue, cell and mouse models.
Patterning
The MGE marker NKX2-1 was expressed within rosettes and in cells rapidly migrating from them, whereas EMX2 was expressed at the base of the rosette where cells were beginning to differentiate (Supplementary Figure 4). Control neurons expressed more EMX2 and less NKX2-1 than BP neurons regardless of culture conditions; PAX6 was expressed in rosettes in both groups. Both BP and control iPSC could be patterned by exposure to the Hedgehog pathway activator purmorphamine by increasing the number of NKX2-1-positive cells (Supplementary Figures 4A and D) or to LiCl by increasing the number of neurons expressing EMX2 (Supplementary Figures 4C and F).
Calcium signaling
By 12 weeks, spontaneous neuronal firing could be observed in these cultures. Lithium pretreatment of BP neurons significantly decreased their calcium transient (101. 65±31. 64 vs 290. 22±0.52 sec; P⩽0.007) and wave amplitude (0.76±0.07 vs 1.89±0.26 ΔF/F0; P⩽0.004) compared with lithium-exposed control neurons (Supplementary Figure 5). We also observed that clusters of soma were capable of clearing Ca2+ more rapidly than axon- and dendrite-rich regions.
Discussion
There is a clear need for cell-based models of BP to test genetic and epigenetic factors, the role of the environment (particularly stress), to determine when neuronal differentiation is affected, and to identify and test new therapeutic approaches. The recent discovery that four transcription factors can reprogram adult somatic cells into PSCs41 has provided an important tool to study human disease.31,57,58 In addition to interrogating the many hypotheses regarding the etiology of BP, the iPSC will be useful in modeling and comparing with other neuropsychiatric disorders that have developmental or affective phenotypes. The banked fibroblast lines also provide a valuable resource for direct reprogramming of the desired cell or neuronal subtype and for alternative reprogramming technologies.
The expression of factors associated with pluripotency as well as their tri-lineage differentiation capability, support the successful reprogramming of dermal fibroblasts to iPSC. Not surprisingly, BP and control iPSCs did not differ significantly in their transcriptional profiles. Although there were many similarities in gene expression, there were differences that will allow us to begin to examine the pathways previously suggested to be affected in BP and those identified in this study including calcium signaling and microRNA processing.
Neurons differentiated from BP iPSC were significantly different in their gene expression profile from those derived from control iPSC. BP neurons expressed more membrane receptors and ion channel genes than control neurons, particularly transcripts involved in calcium signaling. Subtle dysregulation of calcium signaling has been suggested to generate inappropriate neuronal responses and enhance the tonic excitability that maintains brain rhythms in BP.59 Calcium signaling also has a critical role in development, synaptic plasticity and homeostasis of the CNS; misregulation during development can produce subtle but widespread alterations in lineage differentiation and plasticity throughout the nervous system and may influence susceptibility to BP.60 Our preliminary analyses suggest that calcium signaling is also altered in BP neurons and is sensitive to lithium pretreatment. Lithium acts both pre- and post-synaptically, can alter calcium uptake and intracellular levels, and via GSK3β can be neuroprotective. It will now be possible to examine the detailed effects of lithium exposure as well as effects of other BP drugs and other signaling pathways in these cells.
These results support the concept that many complex, neuropsychiatric disorders that are identified later in life, including BP,61, 62, 63 may result from alterations in neural differentiation that occur during development. BP neurons expressed transcripts involved in the differentiation of ventral neuronal subtypes, including FOXP2 and NKX2-1, the hallmark of the medial ganglionic eminence,51 whereas controls expressed higher levels of factors that confer (or maintain) dorsal telencephalic neuronal identity, including EMX2, PAX6, TBR2, TCF3 and ZNF536. As telencephalic neurons are involved in a number of neurodegenerative (Huntington’s disease and Alzheimer’s disease) and neurodevelopmental conditions (autism spectrum disorder, schizophrenia, BP and Rett syndrome), alterations in subtype specification may begin to explain the origin or susceptibility to these conditions.64 Because lithium activates WNT pathway signaling, dorsalizing early CNS progenitors,26 lithium treatment may re-direct neural stem cell fate to dorsal cortical derivatives. Many additional mechanisms may be involved, for example, FOXP2 (expressed at high levels in BP, but not control neurons) binds the DISC1 promoter,65 thereby potentially affecting cell migrations and cortical layering. There were no obvious differences in the expression of transcripts for Hedgehog, Wnt or BMP family members between groups, but the significant increase in transcripts encoding transforming growth factor-β family members in control iPSC may suggest a mechanism underlying the observed differences in dorsal/ventral neuronal phenotypes between groups.
Immunohistochemistry demonstrated an increase in the number of BP neurons expressing the MGE marker NKX2-1, whereas EMX2 was expressed by more control neurons. iPSC remained sensitive to patterning factors as demonstrated by treatment with the Hedgehog pathway activator purmorphamine or to LiCl. Interestingly, preliminary data also demonstrate that this early patterning is maintained with differentiation, with BP neurons expressing higher levels of somatostatin than controls. Considerable research remains to be done to determine the timing and extent to which these alterations are maintained.
The observation that DICER1 was significantly increased in BP neurons suggests a potential mechanism to explain the growing consensus that multiple genes may be misregulated in BP. As microRNAs alter the translation of as many as 70% of all human genes,66 alterations in microRNA production would affect multiple gene products. MicroRNA levels are altered in postmortem brain tissue of BP patients,67,68 and are sensitive to lithium.69, 70, 71, 72 MicroRNAs have critical roles in neural development,73, 74, 75 as well as in the adult brain;76 misregulation would therefore be expected to produce both short- and longer term affects on CNS development and function.
Current work is in progress to expand the patient sample, to examine microRNA expression, to determine the extent to which patterning can be influenced by growth factors42,43 or by the complex three-dimensional microenvironment of the differentiating and mature brain, with the longer term goals of developing new animal models, and ultimately to identify new therapeutic approaches to this intractable disorder.
References
Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ . Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet 2006; 367: 1747–1757.
Murray CJ, Lopez AD . Evidence-based health policy—lessons fro the Global Burden of Disease Study. Science 1996; 274: 740–743.
McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A . The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry 2003; 60: 497–502.
Barnett JH, Smoller JW . The genetics of bipolar disorder. Neuroscience 2009; 164: 331–343.
Liu Y, Blackwood DH, Caesar S, de Geus EJ, Farmer A, Ferreira MA et al. Wellcome Trust Case-Control Consortium. Meta-analysis of genome-wide association data of bipolar disorder and major depressive disorder. Mol Psychiatry 2011; 16: 2–4.
Gershon ES, Alliey-Rodriguez N, Liu C . After GWAS: searching for genetic risk for schizophrenia and bipolar disorder. Am J Psychiatry 2011; 168: 253–256.
Alsabban S, Rivera M, McGuffin P . Genome-wide searches for bipolar disorder genes. Curr Psychiatry Rep 2011; 13: 522–527.
Segurado R, Detera-Wadleigh SD, Levinson DF, Lewis CM, Gill M, Nurnberger JI Jr et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: bipolar disorder. Am J Hum Genet 2003; 73: 49–62.
Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome wide analysis. The Lancet 2013; 381: 1371–1379.
Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 2008; 40: 1056–1058.
Green EK, Grozeva D, Jones I, Jones L, Kirov G, Caesar S et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry 2011; 15: 1016–1022.
Green EK, Hamshere M, Forty L, Gordon-Smith K, Fraser C, Russell E et al. Replication of bipolar disorder susceptibility alleles and identification of two novel genome-wide significant associations in a new bipolar disorder case-control sample. Mol Psychiatry 2012; 18: 1302–1307.
Perrier E, Pompei F, Ruberto G, Vassos E, Collier D, Frangou S . Initial evidence for the role of CACNA1C on subcortical brain morphology in patients with bipolar disorder. Eur Psychiatry 2011; 26: 135–137.
Poliak S, Peles E . The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci 2003; 4: 968–980.
Schulze TG, Detera-Wadleigh SD, Akula N, Gupta A, Kassem L, Steele J et al. Two variants in Ankyrin 3 (ANK3) are independent genetic risk factors for bipolar disorder. Mol Psychiatry 2009; 14: 487–491.
Zhang X, Lei K, Yuan X, Wu X, Zhuang Y, Xu T, Xu R, Han M . SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron 2009; 64: 173–187.
Soeiro-de-Souza MG, Bio DS, Dias VV, Vieta E, Machado-Vieira R, Moreno RA . The CACNA1C risk allele selectively impacts on executive function in bipolar type I disorder. Act Psychiatr Scand 2013; 128: 362–369.
Bearden CE, Hoffman KM, Cannon TD . The neuropsychology and neuroanatomy of bipolar affective disorder: a critical review. Bipolar Disord 2001; 3: 106–150.
DelBello MP, Adler CM, Strakowski SM . The neurophysiology of childhood and adolescent bipolar disorder. CNS Spectr 2006; 11: 298–311.
Kloos A, Weller EB, Weller RA . Biologic basis of bipolar disorder in children and adolescents. Curr Psychiatry Rep 2008; 10: 98–103.
Vawter MP, Freed WJ, Kleinman JE . Neuropathology of bipolar disorder. Biol Psychiatry 2000; 48: 486–504.
Beckmann H, Jakob H . Prenatal disturbances of nerve cell migration in the entorhinal region: a common vulnerability factor in functional psychoses? J Neural Transm Gen Sect 1991; 84: 155–164.
Laeng P, Pitts RL, Lemire AL, Drabik CE, Weiner A, Tang H et al. The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. J Neurochem 2004; 91: 238–251.
Schloesser RJ, Chen G, Manji HK . Neurogenesis and neuroenhancement in the pathophysiology and treatment of bipolar disorder. Int Rev Neurobiol 2007; 77: 143–178.
Mehler MF, Gokhan S . Developmental mechanisms in the pathogenesis of neurodegenerative diseases. Prog Neurobiol 2001; 63: 337–363.
Wang H, Lei Q, Oosterveen T, Ericson J, Matise MP . Tcf/Lef repressors differentially regulate Shh-Gli target gene activation thresholds to generate progenitor patterning in the developing CNS. Development 2011; 138: 3711–3721.
Durnaoglu S, Genc S, Genc K . Patient-specific pluripotent stem cells in neurological diseases. Stem Cells Int 2011; 2011: 212487.
Gaspard N, van der Haeghen P . From stem cells to neural networks: recent advances and perspectives for neurodevelopmental disorders. Dev Med Child Neurol 2010; 53: 13–17.
Ebert AD, Yu J, Rose FF Jr, Mattis VB, Lorson CL, Thomson JA et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009; 457: 277–280.
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012; 482: 216–220.
Juopperi TA, Song H, Ming GL . Modeling neurological diseases using patient-derived induced pluripotent stem cells. Future Neurol 2011; 6: 363–373.
Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, Rhee D et al. Directed conversion of Alzheimer’s disease patient skin fibroblasts into functional neurons. Cell 2011; 3: 359–371.
Urbach A, Bar-Nur O, Daley GQ, Benvenisty N . Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010; 6: 407–411.
Zhang J, Lian Q, Zhou F, Sui L, Tan C, Mutalif RA et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011; 8: 31–45.
Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011; 473: 221–225.
Lin M, Hrabovsky A, Pedrosa E, Wang T, Zheng D, Lachman HM . Allele-biased expression in differentiating human neurons: implications for neuropsychiatric disorders. PLoS ONE 2012; 7: e44017.
Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med 2011; 17: 1657–1662.
Robicsek O, Karry R, Petit I, Salman-Kesner N, Muller FJ, Klein E et al. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenic patients. Mol Psychiatry 2013; 18: 1067–1076.
Langenecker SA, Saunders EF, Kade AM, Ransom MT, McInnis MG . Intermediate cognitive phenotypes in bipolar disorder. J Affect Disord 2010; 122: 285–293.
Nurnberger JI, Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J et al. The diagnostic interview for genetic studies: Rationale, unique features, and training. Arch Gen Psychiatry 1994; 51: 849–859.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K et al. Induction of pluripotent stem cells by defined factors. Cell 2007; 131: 861–872.
Maroof AM, Keros S, Tyson JA, Ying S-W, Ganat YM, Merkle FT et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 2013; 12: 559–572.
Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 2013; 12: 573–586.
Huang DW, Sherman BT, Lempicki RA . Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc 2009; 4: 44–57.
Rajapakse I, Perlman MD, Scalzo D, Kooperberg C, Groudine M, Kosak ST . The emergence of lineage-specific chromosomal topologies from coordinate gene regulation. Proc Natl Acad Sci USA 2009; 106: 6679–6684.
Pfaffl MW, Horgan GW, Dempfle L . Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 2012; 30: e36.
Lee P, Klos M, Bollensdorff C, Hou L, Ewart P, Kamp TJ, Zhang J et al. Simultaneous voltage and calcium mapping of genetically purified human induced pluripotent stem cell – derived cardiac myocyte monolayers. Circ Res 2012; 110: 1556–1563.
Mangale VS, Hirokawa KE, Satyaki PR, Gokulchandran N, Chikbire S, Subramanian L et al. Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science 2008; 319: 304–309.
Goulburn AL, Alden D, Davis RP, Micallef SJ, Ng ES, Yu QC et al. A targeted NKX2.1 Hesc reporter line enables identification of human basal forebrain derivatives. Stem Cells 2011; 29: 462–473.
Jakovcevski I, Mayer N, Zecevic N . Multiple origins of human neocortical interneurons are supported by distinct expression of transcription factors. Cereb Cortex 2011; 21: 1771–1782.
Nat R Apostolova G Dechang G . Telencephalic neurogenesis versus telencephalic differentiation of pluripotent stem cells. In: Wislet-Gendebien, S. Trends in Cell Signaling Pathways in Neuronal Fate Decision. InTech: Rijeka, Croatia, 2013.
Sundberg M, Andersson PH, Akesson E, Odeberg J, Holmberg L, Inzunza J et al. Markers of pluripotency and differentiation in human neural precursor cells derived from embryonic stem cells and CNS tissue. Cell Transplant 2011; 20: 177–191.
Smith JR, Vallier L, Lupo G, Alexander M, Harris WA, Pedersen RA . Inhibition of activin/nodal signaling promotes specification of human embryonic stem cells into neuroectoderm. Dev Biol 2008; 313: 107–117.
Uezato A, Meador-Woodruff JH, McCullumsmith RE . Vesicular glutamate transporter mRNA expression in the medial temporal lobe in major depressive disorder, bipolar disorder, and schizophrenia. Bipolar Disord 2009; 11: 711–725.
Cecil KM, DelBello MP, Sellars MC, Strakowski SM . Proton magnetic resonance spectroscopy of the frontal lobe and cerebellar vermis in children with a mood disorder and a familial risk for bipolar disorders. J Child Adolesc Psychopharmacol 2003; 13: 545–555.
Chen H, Wang N, Zhao X, Ross CA, O’Shea KS, McInnis MG . Gene expression alterations in bipolar disorder brains. Bipolar Disord 2013; 15: 177–187.
Brennand KJ, Simone A, Tran N, Gage FH . Modeling psychiatric disorders at the cellular and network levels. Mol Psychiatry 2012; 17: 1239–1253.
Mattis VB, Svendsen CN . Induced pluripotent stem cells: a new revolution for clinical neurology? Lancet 2011; 10: 383–394.
Berridge MJ . Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 2013; 7: 2–13.
Van Meter AR, Moreira AL, Youngstrom EA . Meta-analysis of epidemiologic studies of pediatric bipolar disorder. J Clin Psychiatry 2011; 72: 1250–1256.
Howes OD, Falkenberg I . Early detection and intervention in bipolar affective disorder: targeting the development of the disorder. Curr Psychiatry Rep 2011; 13: 493–499.
Nasrallah HA . Neurodevelopmental aspects of bipolar affective disorder. Biol Psychiatry 1991; 29: 1–2.
Sigurdsson E, Fombonne E, Sayal K, Checkley S . Neurodevelopmental antecedents of early-onset bipolar affective disorder. Br J Psychiat 1999; 174: 121–127.
Marin O . Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci 2012; 13: 107–120.
Walker RM, Hill AE, Newman AC, Hamilton G, Torrance HS, Anderson SM et al. The DISC1 promoter: characterization and regulation by FOXP2. Hum Mol Genet 2012; 21: 2862–2872.
Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006; 126: 1203–1217.
Kim AH, Reimers M, Maher B, Williamson V, McMichael O, McClay JL et al. MicroRNA expression profiling in the prefrontal cortex of individuals affected with schizophrenia and bipolar disorders. Schizophr Res 2010; 124: 183–191.
Moreau MP, Bruse SE, David-Rus R, Buyske S, Brzustowicz LM . Altered microRNA expression profiles in postmortem brain samples from individuals with schizophrenia and bipolar disorder. Biol Psychiatry 2011; 69: 188–193.
Chen H, Wang N, Burmeister M, McInnis MG . MicroRNA expression changes in lymphoblastoid cell lines in response to lithium treatment. Int J Neuropsychopharmacol 2009; 12: 975–981.
Rong H, Liu TB, Yang KJ, Yang HC, Wu DH, Liao CP et al. MicroRNA-134 plasma levels before and after treatment for bipolar mania. J Psychiatr Res 2011; 45: 92–95.
Zhou R, Yuan P, Wang Y, Hunsberger JG, Elkahloun A, Wei Y et al. Evidence for selective microRNAs and their effectors as common long-term targets for the actions of mood stabilizers. Neuropsychopharm 2009; 34: 1395–1405.
Weinberger DR . Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 1987; 44: 660–669.
Kritchevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS . A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 2003; 9: 1274–1281.
Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Sestan N, Rakic P et al. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol 2004; 5: R68.
Smirnova L, Grafe A, Seiler A, Schumacher S, Nitsch R, Wulczyn FG . Regulation of miRNA expression during neural cell specification. Eur J Neurosci 2005; 21: 1469–1477.
Cheng HY, Papp JW, Varlamova O, Dziema H, Russell B, Curfman JP et al. microRNA modulation of circadian-clock period and entrainment. Neuron 2007; 54: 813–829.
Acknowledgements
We are grateful to the Heinz C. Prechter Bipolar Research Fund, the Steven M. Schwartzberg Memorial Fund, The Joshua Judson Stern Foundation and the A. Alfred Taubman Medical Research Institute for support. MGM is the Thomas B and Nancy Upjohn Woodworth Professor of Bipolar Disorder and Depression. We thank Dr Johann Gudjonsson for obtaining skin biopsies and Dr Gary Smith for advice with the research. We are grateful to the research participants in the Prechter Longitudinal Bipolar Study and the many research associates and administrative staff at the University of Michigan for attending to details critical to the success of the team. Studies were reviewed and approved by the University of Michigan human Pluripotent Stem Cell Research Oversight (hPSCRO) committee and the IRBMed.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Chen, H., DeLong, C., Bame, M. et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl Psychiatry 4, e375 (2014). https://doi.org/10.1038/tp.2014.12
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.12
Keywords
This article is cited by
-
Progress and Implications from Genetic Studies of Bipolar Disorder
Neuroscience Bulletin (2024)
-
APOE deficiency impacts neural differentiation and cholesterol biosynthesis in human iPSC-derived cerebral organoids
Stem Cell Research & Therapy (2023)
-
Bipolar disorder-iPSC derived neural progenitor cells exhibit dysregulation of store-operated Ca2+ entry and accelerated differentiation
Molecular Psychiatry (2023)
-
Advances and Applications of Brain Organoids
Neuroscience Bulletin (2023)
-
CADPS functional mutations in patients with bipolar disorder increase the sensitivity to stress
Molecular Psychiatry (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}