
Abstract
Anhedonia—which is defined as diminished pleasure from, or interest in, previously rewarding activities—is one of two cardinal symptoms of a major depressive episode. However, evidence suggests that standard treatments for depression do little to alleviate the symptoms of anhedonia and may cause reward blunting. Indeed, no therapeutics are currently approved for the treatment of anhedonia. Notably, over half of patients diagnosed with bipolar disorder experience significant levels of anhedonia during a depressive episode. Recent research into novel and rapid-acting therapeutics for depression, particularly the noncompetitive N-Methyl-D-aspartate receptor antagonist ketamine, has highlighted the role of the glutamatergic system in the treatment of depression; however, it is unknown whether ketamine specifically improves anhedonic symptoms. The present study used a randomized, placebo-controlled, double-blind crossover design to examine whether a single ketamine infusion could reduce anhedonia levels in 36 patients with treatment-resistant bipolar depression. The study also used positron emission tomography imaging in a subset of patients to explore the neurobiological mechanisms underpinning ketamine’s anti-anhedonic effects. We found that ketamine rapidly reduced the levels of anhedonia. Furthermore, this reduction occurred independently from reductions in general depressive symptoms. Anti-anhedonic effects were specifically related to increased glucose metabolism in the dorsal anterior cingulate cortex and putamen. Our study emphasizes the importance of the glutamatergic system in treatment-refractory bipolar depression, particularly in the treatment of symptoms such as anhedonia.
Similar content being viewed by others

Introduction
Over half of patients diagnosed with bipolar disorder (BD) suffer from significant levels of anhedonia,1 defined as loss of enjoyment in, or desire to engage in, previously pleasurable activities.2 Notably, anhedonic patients with affective disorders have a poorer treatment prognosis than their non-anhedonic counterparts.3, 4, 5 Indeed, accumulating evidence suggests that standard treatments for depression do little to alleviate anhedonia6 and may even cause reward and emotional blunting,7, 8, 9 sexual anhedonia10 and anorgasmia.11,12 Furthermore, the presence of anhedonia in a major depressive episode (MDE) is a predictor of proximal suicide completion.13 Critically, no US Food and Drug Administration-approved treatment currently exists specifically for anhedonia.
The Diagnostic and Statistical Manual-5 (ref. 14) identifies anhedonia as one of two cardinal symptoms in the diagnosis of an MDE in both major depressive disorder (MDD) and BD. Anhedonia can be subdivided into consummatory (subjective pleasure, for example, enjoying food) and motivational components (anticipation of and drive towards rewarding stimuli, for example, planning and looking forward to a vacation) that have distinct biological bases.15
Indeed, research suggests that currently depressed MDD and BD patients may possess a substantial deficit in motivational, but not consummatory, reward behaviors.16,17 Studies using the sweet taste test—a task that mirrors preclinical assessments of consummatory anhedonia in rodents—found that patients with MDD demonstrated the same preference for sucrose water concentrations as healthy volunteers.18,19 Furthermore, Sherdell et al.20 found that MDD patients experienced the same levels of pleasure as healthy volunteers while viewing humorous cartoons in a computer task, but were not willing to exert as much effort to gain access to these stimuli; the results suggest intact consummatory processes, but attenuated motivational ones. In another study, Etain et al.21 found no evidence for consummatory anhedonia in BD patients. Although research pertaining to BD patients in particular is lacking, overall, the extant evidence suggests that anhedonia in depression is primarily associated with a deficit in non-consummatory reward behaviors. Understanding the neural pathways that mediate anticipatory pleasure is thus a critical step towards successful treatment of anhedonia.
Dopaminergic signaling has been consistently correlated with the anticipation, motivation and learning related to pleasurable stimuli, but not to their consumption.22, 23, 24 Phasic bursts in dopaminergic neurons in the ventral tegmental area (VTA) have reliably been shown to co-occur with violations in reward expectancy,25 underscoring the evidence for dopaminergic signalling in reward learning. Furthermore, dopamine signalling in the nucleus accumbens, an area of dense dopaminergic projections from the VTA, has been strongly associated with reward motivation in rodents.26 Functional neuroimaging in humans indicates that structures such as the VTA,27,28 substantia nigra,28 amygdala,28 putamen,28 caudate,28 ventral striatum,29,30 and orbitofrontal cortex28,31—all of which receive innervation from or project to dopaminergic nuclei—are recruited during reward anticipation.
Despite the abnormalities in motivational behaviors seen in affective disorders, there is a dearth of robust evidence pertaining to any direct dopaminergic signaling deficit in patients with depression.32 The strongest indirect evidence for dopaminergic dysfunction in depression comes from pharmacological treatment studies. For instance, the dopamine D2 receptor agonist pramipexole improved levels of depression in both MDD33 and BD34,35 patients after several weeks of daily administration. A study of Parkinson’s disease patients with co-occurring depression used the Snaith–Hamilton Pleasure Scale (SHAPS36), and found that pramipexole treatment decreased anticipatory anhedonia levels by 25% across the entire sample;37 however, it is unknown whether this reduction occurred as a function of improvement in mood, Parkinson’s symptomatology or hedonic capacity.
It is presently unclear whether anhedonic symptoms in depression improve faster with dopaminergic-enhancing medications than with standard treatments. Furthermore, because improvements in self-reported levels of anhedonia are reportedly the last symptom to improve with selective serotonin reuptake inhibitors,38 there is a critical unmet need for rapid-acting treatments for anhedonia. The fact that standard antidepressants lack any proven anti-anhedonic efficacy, particularly in conjunction with the deleterious side effects associated with these agents, requires novel pharmacotherapeutic approaches.
The noncompetitive N-Methyl-D-aspartate (NMDA) receptor antagonist ketamine has shown remarkable consistency in rapidly ameliorating depressive symptoms in both MDD39, 40, 41, 42, 43 and BD.44,45 However, it is unknown whether ketamine also possesses any specific anti-anhedonic efficacy. Given the likely mechanistic heterogeneity of depression, it is critical to understand the specific targets of treatment response at both the clinical and neural levels, as outlined in the research domain criteria.46 Ketamine acts directly on the glutamatergic system, which appears to be critical in depression;47,48 however, little is known about the specificity of the relationship between commonly occurring symptoms during an MDE (for instance, anhedonia, anxiety) and particular biological phenotypes. In a small sample investigation, Walter et al.49 found that MDD patients with high levels of anhedonia had lower levels of glutamine, but not glutamate, than healthy controls, but only a trend towards lower glutamine than MDD patients with low levels of anhedonia, who did not differ from controls. Preclinical evidence suggests that blockade of astrocytic glutamate reuptake in rodents can induce anhedonia-like behaviors,50 particularly in the prefrontal cortex.51 In addition, ketamine52,53 and other NMDA antagonists (for example, MK-80154 and memantine55) have been shown to reverse anhedonic phenotypes in rodents. However, preclinical evidence typically classifies anhedonia as a decrease in consummatory behavior; although anticipatory and consummatory behaviors likely interact, extrapolating from rodent behaviors to clinical patient symptoms is not straightforward. Sub-anesthetic doses of ketamine acutely increase glutamatergic and dopaminergic signaling in the prefrontal cortex of rats.56 Given the apparent role of dopaminergic and glutamatergic signaling in mediating anhedonia and the reported pharmacological effects in both rodents and humans, ketamine may be ideally suited to specifically ameliorate anticipatory anhedonia in currently depressed patients.
This randomized, double-blind, placebo-controlled, crossover study assessed the anti-anhedonic efficacy of ketamine in treatment-resistant BD patients currently experiencing an MDE. Regional neural metabolism following both placebo and ketamine infusions were also measured in a subsample of these patients using [18F] fluorodeoxyglucose (FDG) positron emission tomography (PET). 18FDG-PET measures glucose metabolism, which is primarily determined by glial uptake of glucose in response to glutamate release from neurons, and principally reflects glutamatergic neurotransmitter release and cycling.57 It is important to note that approximately all glucose entering the central nervous system is transformed into glutamate.58 Thus, 18FDG-PET provides an indirect quantitative measure of cerebral glutamate metabolism throughout the entire brain. The effects of ketamine on general depressive symptoms in the majority of BD subjects presented here (33/36, 92%) were previously reported,44,45 together with neurobiological correlates of mood improvement derived from 18FDG-PET.59 The present study specifically explores the previously unaddressed issue of ketamine’s anti-anhedonic effects and its neural correlates in this sample.
Materials and methods
Participants
Subjects aged 18–65 were recruited through local and national media. The sample comprised 36 (21F) treatment-refractory individuals with BD I or II without psychotic features who were currently experiencing an MDE (see Table 1). All subjects were inpatients at the National Institutes of Health (NIH), Bethesda, MD, USA. A Montgomery–Åsberg Depression Rating Scale (MADRS)60 score ⩾20 at the time of screening was an inclusion criterion. All subjects were required to be currently experiencing an MDE lasting at least 4 weeks and to have failed to respond to at least one adequate antidepressant trial before hospital admission, as assessed by the Antidepressant Treatment History Form.61 Diagnosis was confirmed using the Structured Clinical Interview for Axis I Diagnostic and Statistical Manual-IV Disorders-Patient Version.62 Primary psychiatric categorization, including comorbid Axis I disorders, was corroborated via evaluation by three clinicians using all available clinical information. During their time as inpatients at the NIH, and before study entry, participants had also not responded to a prospective open-label trial of a mood stabilizer (either lithium or valproic acid) administered at treatment levels (serum lithium, 0.6–1.2 mEq l−1; or valproic acid, 50–125 μg ml−1).
All patients were in good physical health as determined by medical examination, medical history, chest x-ray, blood laboratory tests, urinary analysis and toxicology. Exclusion criteria, as outlined previously,44,45 included pregnancy, nursing, serious suicidal ideation, comorbid substance abuse or dependence within the past 3 months, and previous use or treatment with ketamine. The co-occurrence of Axis I anxiety disorders was permitted if this was not the primary cause of illness within the previous 12-month period. All participants continued treatment with a mood stabilizer (either lithium or valproic acid) administered at therapeutic levels during the course of the study, although this was not adequate to alleviate depressive symptoms. Aside from monotherapy with a mood stabilizer, no psychotropic medication or psychotherapy was permitted in the 2 weeks before study randomization (5 weeks for fluoxetine) or during the 4-week study period. Written informed consent was obtained from all participants, and the NIH Combined Neuroscience Institute Review Board approved the study.
Design
The study was designed as a randomized, double-blind, placebo-controlled, crossover study to assess the antidepressant efficacy of ketamine in patients with treatment-refractory BD. All participants received one intravenous infusion of ketamine hydrochloride, administered at a subanesthetic dose of 0.5 mg kg−1, and one infusion of placebo (0.9% saline solution) in a randomized order over a 4-week study period, and with 2 weeks between each infusion. A MADRS score ⩾20 on the morning of each infusion was required for study continuation. An anesthesiologist administered each infusion over 40 min using a Baxter infusion pump (Deerfield, IL, USA). The two solutions and the appearance of the drug syringe were identical and all study team members were blind to the drug condition.
Clinical ratings were acquired 60 min before the infusion and thereafter at 40, 80, 120, 230 min, and days 1, 2, 3, 7, 10 and 14 following each infusion. The primary outcome variable for antidepressant efficacy was the MADRS44,45 score. However, the main symptom of interest for this report is anhedonia, and levels of anticipatory anhedonia were evaluated using the SHAPS. The SHAPS is a 14-item, self-administered, user friendly and state-sensitive psychometric scale that overcomes limitations associated with other scales that specifically assess anhedonia, such as length and cultural idiosyncrasies. Importantly, the SHAPS has been validated in a number of independent samples since its publication.63, 64, 65 The SHAPS was scored on a scale of one to four, with higher scores indicating greater anhedonia (range 14–56) and was administered with reference to either the past 24 h or the time between the present and the previous rating. The presence or absence of anhedonia was judged on the basis of the original scoring guidelines (range 0–14),36 where a score >3 indicated clinically significant anhedonia (see Table 1). Other secondary outcome variables were also assessed and have been reported previously.44,45
Positron emission tomography acquisition and analysis
In addition, 21 of the 36 patients (see Table 1) underwent two resting-state 18FDG-PET imaging scans, which began 2 h post infusion and ended (brain emission scan) ~1.5 h later. Immediately before both scans, patients completed psychometric rating scales; identical procedures were followed for both scans. PET imaging was performed on a GE Advance PET scanner (GE Medical Systems, Waukesha, WI, USA) in three-dimensional mode (35 contiguous slices, 4.25 mm plane separation) following an intravenous infusion of 4.5 mCi 18FDG over 2 min. According to the method used by Brooks,66 quantitative images of regional cerebral metabolic rate for glucose metabolism (rCMRGlu) were calculated using a cardiac input function derived from a dynamic left ventricular scan collected before both the brain emission scan and venous sampling (reconstructed resolution=6 mm full-width at half-maximum in all planes). Magnetic resonance imaging (MRI) images were acquired on a 3-Tesla scanner (Signa, GE Medical Systems) using a three-dimensional MPRAGE sequence (echo time=2.982 ms, repetition time=7.5 ms, inversion time=725 ms, voxel size=0.9 × 0.9 × 1.2 mm) to allow anatomic localization of PET activity. PET analyses comprised both a region of interest approach (ROI) and a whole-brain investigation.
The ROI analysis pipeline used here has been previously described.59 Briefly, the Analysis of Functional NeuroImages (AFNI; Bethesda, MD, USA) function 3dSkullStrip was used to remove non-brain tissue from the anatomical MRI image. These images were segmented into gray and white matter, and cerebrospinal fluid (separate binary mask images were computed for each component) using the FSL (Oxford, UK) automated segmentation tool. Anatomical images were spatially normalized to the Montreal Neurological Institute 152 template. ROIs (ventral striatum and orbitofrontal cortex) were selected on the basis of extant literature implicating these neuroanatomical structures in depression67, 68, 69, 70 and reward anticipation.16,31 The ventral striatum ROI comprised only the nucleus accumbens and olfactory tubercle. Template-defined ROIs were transferred to the individual anatomical MRIs; to accommodate interindividual anatomical variation, ROI placement was adjusted per subject. ROIs were transferred back to the native MRI space, multiplied by a binary gray matter mask and applied to the rCMRGlu PET images. Mean glucose metabolism rate values, normalized by total gray matter, were then calculated.
For the whole-brain investigation, 18FDG-PET images were preprocessed and analyzed using Statistical Parametric Mapping software (SPM; Wellcome Trust Centre for Neuroimaging, London, UK) version 5 within the MATLAB (MathWorks, Natick, MA, USA) environment. Post-placebo and post-ketamine rCMRGlu images were separately co-registered to the anatomical image; the anatomical image was then normalized to Montreal Neurological Institute space and this transformation was applied to the co-registered PET images. A Gaussian smoothing kernel (8 mm full-width at half-maximum) was applied to the PET images. To create difference images for each individual, PET images were first normalized by the global mean (as calculated in SPM) and the post-placebo image was subtracted from the post-ketamine image. A binary mask was applied to all whole-brain investigations to limit the number of multiple comparisons to only intracerebral voxels.
Statistical analyses
Symptom rating scale analysis included all available data and was conducted using IBM SPSS (Armonk, NY, USA; version 21). Linear mixed models, using a heterogeneous first-order autoregressive covariance structure and restricted maximum-likelihood estimation, were performed to assess the effects of ketamine versus placebo on SHAPS scores over the 4-week period in this crossover design. Fixed main effects of time and drug and their interaction were included along with a random effect for subject. To correct for baseline levels of anhedonia, the SHAPS score at baseline on each infusion day (time point −60) was entered as a covariate into the model. In addition, a linear mixed model with total MADRS score (at each individual time point) entered as a covariate was conducted to evaluate whether ketamine infusion was associated with a change in SHAPS score independent of the effect on other depressive symptoms. MADRS item 8 (inability to feel) was removed from the total MADRS score for this analysis due to the strong conceptual overlap between this item and the SHAPS. Post hoc Bonferonni-corrected comparisons were conducted for each model for all post-baseline time points to determine the specific timing of the anti-anhedonic effects. All significance values were two-tailed, with a significance threshold of P<0.05.
The time point of 230 min was a priori selected for both the ROI and whole-brain analyses on the basis of three factors: previous studies indicating that 230 min is a sensitive time point for detecting antidepressant effects of ketamine;44,45 lack of psychotomimetic effects at this time point; and the proximity to the time of the PET scan. Relationships between glucose metabolism in our ROIs (post-placebo and post-ketamine and their difference) and SHAPS score were investigated using Pearson product moment correlation coefficients. First, percentage improvement in SHAPS score at 230 min post infusion ((post-ketamine 230−post-placebo 230)/post-placebo 230) was correlated with difference in mean ROI rCMRGlu metabolism (post-ketamine–post-placebo). Second, because we previously demonstrated an association between ventral striatum metabolism changes and overall depression score change following ketamine,59 we conducted a multiple linear regression analysis to parse the variance associated with anhedonia and total depression score and explore which variable predicted change in ventral striatum metabolism. Finally, as a control analysis, we also assessed whether state-dependent anhedonia levels, as measured by raw SHAPS score, were associated with ROI rCMRGlu, both variables post-ketamine and post-placebo.
Complementing the ROI analysis, the whole-brain investigation comprised the following multiple regression analyses. First, percent improvement on the SHAPS at 230 min (as above) was regressed onto the difference images (post-placebo–post-ketamine). Second, to assess the specificity of the results to anhedonia, and not depressive symptoms per se, we recomputed the whole-brain analyses with percentage change anhedonia scores orthogonalized to the corresponding percentage change in MADRS score (minus item 8) using the SPM spm_orth function within the MATLAB environment, entering a single regressor (SHAPS score orthogonalized to total MADRS score minus item 8). Orthogonalization of one variable against another, in this instance the SHAPS against the MADRS score, results in the removal of shared variance; the output variable represents the residual SHAPS score when the variance associated with the MADRS has been accounted for. Here, we report only those analyses that survived stringent Gaussian random field theory cluster correction for multiple comparisons at P<0.05, or at trend level at P<0.1, with an initial voxel-level threshold of P<0.05 uncorrected. In the case of extremely large clusters, the uncorrected threshold was increased so that all surviving clusters were <5000 contiguous voxels. For clusters surviving correction or trending, the three strongest peaks (t statistics) are reported along with the corresponding Montreal Neurological Institute coordinates.
Results
Subjects
Patient demographic details are presented in Table 1. One patient was excluded from the PET analyses due to a failure to measure the cardiac input function, and another subject was excluded because there was no SHAPS scale score measurement at 230 min post-ketamine and post-placebo infusions.
Behavioral response
Main effects of drug (F(1,110)=24.71, P<0.001) and time (F(9,194)=2.69, P=0.006) were observed, as was a trend towards an interaction between these two variables (F(9,218)=1.90, P=0.053); this indicates that ketamine caused a greater reduction in levels of anhedonia across time than placebo, (Figure 1a). Bonferonni-corrected post hoc comparisons demonstrated that ketamine, compared with placebo, significantly decreased levels of anhedonia at multiple times throughout the 14-day period following a single ketamine infusion (Figure 1a).

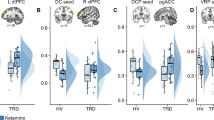
Anti-anhedonic effect of ketamine and corresponding regression analyses with cerebral glucose metabolism. (a) Snaith–Hamilton Pleasure Scale (SHAPS) estimated scores from linear mixed model 1 (M1) indicating a significant reduction in anhedonia levels following ketamine (red) compared with placebo (blue). (b) Model 2 (M2) is the same as model 1 (M1) but has total depression score (as assessed by the Montgomery–Åsberg Depression Rating Scale (MADRS) minus item 8) entered as a covariate and still reveals a main effect of drug, thus underscoring the unique anti-anhedonic effect of ketamine administration. Asterisks indicate Bonferonni-corrected comparisons at P<0.05 for both A and B. (c) Region of interest analysis with ventral striatum (VS) demonstrating a significant association between anti-anhedonic response to ketamine and increased glucose metabolism in the VS. Changes in anhedonia levels no longer significantly predicted VS change when change in overall depressive symptoms were controlled for. (d, e) Whole-brain corrected relationship between the anti-anhedonic effects of ketamine and (d) dorsal anterior cingulate cortex (dACC), cerebellum, (e) right putamen, VS and medial posterior orbitofrontal cortex increases in glucose metabolism. (f) Whole-brain corrected relationship between SHAPS score orthogonalized against MADRS score indicating that a significant increase in dACC metabolism was associated specifically with anti-anhedonic response to ketamine independent of overall change in depressive symptoms. Error bars represent standard errors. PET images are presented (d and e, P<0.025 uncorrected; f, P<0.05 uncorrected) such that only clusters surviving family-wise error correction are shown. Color bars indicate positive t-values associated with increasing glucose metabolism. ket, ketamine; pla, placebo; rCMRGlu, regional cerebral metabolic rate for glucose metabolism.
Consistent with previous reports,1 levels of anhedonia (as measured by the SHAPS) and depressive symptoms (as measured by total MADRS score) were positively correlated (r(36) =0.53, P<0.001) at the first pre-infusion baseline. Critically, the main effect of drug on anhedonia levels was found when levels of depression (MADRS total score minus item 8 at each time point) were entered as a covariate in the model (F(1,123)=7.713, P =0.006), indicating that anhedonia levels in BD respond to ketamine treatment over-and-above its effects on other depressive symptoms. Neither the main effect of time (F(9,176)=1.42, P=0.18) nor the drug-by-time interaction (F(9,219)=1.49, P=0.15) were significant for this model. However, post-hoc exploratory simple effects tests (Bonferonni-corrected) revealed that anti-anhedonic effects of ketamine were significant at days 1, 3, 7 and 14 following ketamine infusion (Figure 1b). This suggests that for some BD patients, ketamine may have specific benefits in reducing anhedonia levels, and that these benefits can last up to 2 weeks following a single infusion.
A subsequent model included mood stabilizer as an additional covariate; a trend towards significance, with greater anti-anhedonic response associated with lithium than valproate was observed (F(1,57)=3.57, P=0.06). No interaction effect was noted between drug (ketamine or placebo) and mood stabilizer (F(1, 114)=0.08, P=0.78), nor was there a three-way interaction between drug, mood stabilizer and time (F(9, 225)=0.41, P=0.93).
Given our previous findings that a positive family history of alcohol use disorder (1st degree relative) was associated with an enhanced response to ketamine,71,72 we examined whether this factor was also associated with the specific anti-anhedonic response demonstrated above; baseline SHAPS score and total depression score were entered as covariates into a subsequent model. There was a significant interaction between drug and family history of alcohol use disorder (F(1,109)=4.12, P=0.045), but no main effect for family history of alcohol use disorder (F(1,51)=0.27, P=0.60), nor an interaction between this variable and time (F(9,163)=0.94, P=0.49), nor a three-way interaction between family history, drug and time (F(9,205)=0.60, P=0.79). The significant interaction between drug and family history of an alcohol use disorder indicated, in comparison with placebo, a specific enhanced anti-anhedonic response to ketamine in those with (F(1,116)=11.13, P =0.001), in comparison with those without (F(1,111)=1.22, P =0.27), a family history of an alcohol use disorder.
In addition, we explored whether a personal history of alcohol abuse, dependence or illicit substance abuse contributed significantly to the specific anti-anhedonic effect of ketamine. No significant main effects or interactions with drug, time or drug and time were found.
PET: ROI analyses
Ketamine-induced change in ventral striatum rCMRGlu was significantly related to percent change in SHAPS score at 230 min post infusion (r(19)=−0.52, P=0.02; Figure 1c). Relative to placebo, individuals with the largest increase in glucose metabolism in the ventral striatum tended to have the highest anti-anhedonic response to ketamine. However, orbitofrontal cortex rCMRGlu activity was not significantly related to anti-anhedonic response to ketamine (r(19)=−0.37, P=0.12). Because we had previously demonstrated a relationship between ventral striatum change in glucose metabolism and improvement in MADRS score following ketamine,59 we tested whether this relationship was specific to anhedonia. Multiple regression, including both SHAPS and MADRS (minus item 8), indicated that change in MADRS (t=−2.18, P=0.045), but not SHAPS (t=−0.05, P=0.96), significantly predicted change in ventral striatum rCMRGlu.
Examining each session separately, we found no significant correlations between absolute SHAPS scores and ventral striatum or orbitofrontal cortex rCMRGlu, respectively, neither post placebo (r(19)=0.03, P=0.91; r(19)=0.10, P=0.68) nor post ketamine (r(20)=0.06, P=0.81; r(20)=0.06, P=0.81).
PET: Whole-brain analyses
Due to the presence of a large cluster, the statistical threshold was raised to 0.025 uncorrected, as opposed to 0.05, for the initial difference image contrast only. Whole-brain analyses revealed a significant relationship between percent improvement (decrease) in SHAPS scores and rCMRGlu increases in the dorsal anterior cingulate cortex (dACC; [x=−6, y=40, z=43]; t(17)=4.39, Pcorr=0.016; Figure 1d). Furthermore, we found a significant increase in cerebellum ([x=−40, y=−48, z=−58]; t(17)=3.94, Pcorr=0.019; Table 2), and a trend towards an increase in striatal, rCMRGlu ([x=14, y=4, z=10]; t(17)=4.28, Pcorr=0.051; Figure 1e), in relation to the percent amelioration in SHAPS scores. Importantly, the striatal cluster extended from the caudate to the putamen and into the ventral striatum (Table 2), corroborating our ROI analysis. The inverse contrast (the relationship between decreases in rCMRGlu and change in SHAPS score) did not yield any whole-brain corrected results.
Because our ROI results indicated that metabolic increases in the ventral striatum appear to be driven by change in total depression score but not levels of anhedonia, we conducted a subsequent whole-brain analysis. In this model, change in SHAPS score was orthogonalized to change in MADRS score, both at 230 min, and this orthogonalized change in SHAPS score was regressed onto the rCMRGlu difference image. This regression revealed that the specific changes in anhedonia levels following ketamine—which were not related to general changes in depressive symptoms—were in fact associated with increased dACC metabolism ([x=−8, y=40, z=28]; t(17)=4.89, Pcorr=0.027; Figure 1f) that extended into the pregenual cingulate/callosal region and the right dorsolateral prefrontal cortex. In addition, a trend was noted towards significantly increased metabolism in the fusiform gyrus ([x=34, y=−28, z=−20]; t(17)=3.74, Pcorr=0.065) that extended into the claustrum and the putamen, but not the ventral striatum. Finally, we assessed whether change in rCMRGlu was associated with longer-term SHAPS response by correlating the difference in dACC (peak voxel; ketamine-placebo) metabolism with change in SHAPS scores at day 14. There was no significant relationship between change in dACC glucose metabolism and the magnitude of the change in SHAPS score at day 14 (r(18)=0.11, P=0.66).
Discussion
Several notable findings emerged from this study investigating the effects of the rapid-acting antidepressant ketamine on anhedonia in currently depressed treatment-resistant BD patients. Foremost among these findings is that ketamine, compared with placebo, rapidly reduced the levels of anhedonia in these patients; this reduction occurred within 40 min of a single ketamine infusion and lasted up to 14 days. Furthermore, we found that anti-anhedonic effects of ketamine remained significant even when controlling for level of depressive symptoms, suggesting that ketamine has a unique role in ameliorating anhedonia levels independent of other depressive symptoms. This study also used 18FDG-PET to examine a subgroup of these patients and quantify the rCMRGlu correlates of change in anhedonia levels associated with ketamine treatment. Our PET results demonstrated that the neurobiology of this specific anti-anhedonic effect was mediated in part by increases in glucose metabolism in the dACC, and tentatively the putamen, but not the ventral striatum as originally hypothesized.
These results are particularly important from a public health perspective. No approved treatments for anhedonia currently exist despite its prevalence across multiple psychiatric disorders. Thus, ketamine’s rapid effects (within 1 h) on anhedonia levels are a crucial clinical finding. Currently available standard treatments for depression, such as selective serotonin reuptake inhibitors, have few positive effects on levels of anhedonia in MDD patients and, indeed, have occasionally deleterious effects;6, 7, 8, 9, 10, 11,73 no research to date has assessed the effects of mood stabilizers on levels of anhedonia in individuals with BD. Our results lend credence to the notion that similar compounds (for example, other NMDA receptor antagonists) should be explored for their clinical relevance in treating this debilitating symptom of depression. The results further suggest that more typically anhedonic subtypes of depression (both MDD and BD)—for instance, melancholic depression—may be particularly suited to treatment with ketamine and its analogs.
Given our previous finding59 that improvement in total depression score on the MADRS was associated with increased ventral striatum rCMRGlu following ketamine, we hypothesized that this region would also be strongly related to anhedonia and its amelioration. Unexpectedly, we found that ventral striatal glucose metabolism was not associated with relative changes in anhedonia levels after controlling for levels of depression. Possible reasons for this result include the severity of illness in these treatment-resistant BD subjects, the underlying biology of the ventral striatum or the distinct symptom assessed by the SHAPS in comparison with the MADRS. Alleviating depressive symptoms is intensely relieving, and thus also rewarding, for treatment-refractory patients. Intriguingly, opioid receptors in the medial accumbens shell are believed to be solely responsible for ‘liking’ or consummatory pleasure behaviors,74 whereas motivational hedonic behaviors are thought to arise from a wide array of receptors within the accumbens—the primary structure in the ventral striatum—as well as other neural structures; 18FDG-PET cannot currently differentiate these structures. Tentatively, we hypothesize that patients showing the greatest antidepressant response to ketamine, as measured by the MADRS, may be experiencing high levels of pleasure, a construct not assessed by the SHAPS. Further investigations are needed to disentangle specific symptoms from the systems level antidepressant mechanisms of action of ketamine in humans.
However, our study did find that individuals with the largest increase in glucose metabolism (post-ketamine–post-placebo) in the dACC and the putamen had the greatest clinical reduction in anhedonia levels; notably, this was true with and without controlling for total depression score. Findings from both human and animal studies indicate that both the dACC and putamen are highly involved in reward processing, learning and decision-making.75, 76, 77, 78 Shidara and Richmond75 demonstrated that reward expectancy, or motivation, was highly correlated with single neuron signals in the monkey ACC (area 24c), a proximal region to the dACC rCMRGlu changes found here. Intriguingly, the dACC has also been strongly linked with the anticipation of rewarding events in humans.79 The dACC rCMRGlu increases seen in the present study may reflect changes in the motivation and anticipation of, or ability to anticipate, pleasurable events; this is reflected in items of the SHAPS (‘I would enjoy a…’). Deficits in the ability to imagine future events, particularly positive ones, have been reliably identified in MDD patients experiencing an MDE;80,81 thus, attenuation of depressive symptoms may be accompanied by an improved ability to anticipate pleasure. In a functional MRI study in humans, O’Doherty et al.28 found that anticipating a primary reward (glucose) elicited heightened activity in the right putamen (as found here) compared with anticipating a punishment (salt water). Keedwell et al.82 found that anhedonia levels, as measured by the Fawcett–Clark Pleasure Scale,83 were positively correlated with activity in the right putamen during an emotional face-processing task that required MDD patients to compare sad and neutral faces; this result was not observed with depression or anxiety scales, though it should be noted that the Fawcett–Clark Pleasure Scale indiscriminately measures both consummatory and anticipatory anhedonia. Taken together, the results of the imaging portion of this study indicate that decreased anticipatory anhedonia is associated with increased glucose metabolism in the dACC and putamen, and that this in turn may reflect increased motivation towards or ability to anticipate pleasurable experiences.
Another interesting finding was that, after the ketamine infusion, individuals taking lithium experienced greater anti-anhedonic effects than those receiving valproate when the antidepressant effect was controlled for. This result could be interpreted in two ways: either valproate caused a deficit in the anti-anhedonic effect of ketamine, or lithium enhanced the anti-anhedonic effect of ketamine. A recent preclinical report by Liu et al.84 found that a single subanesthetic dose of ketamine in conjunction with lithium resulted in enhanced plasticity and antidepressant-like effects (increased struggling time in the forced swim test) in rodents compared with a single dose of ketamine plus vehicle. In light of this result, we suggest that lithium may enhance the anti-anhedonic effects of ketamine in individuals with BD. Future studies in patients both taking and not taking mood stabilizers will need to be conducted to correctly evaluate this possibility.
Ketamine acts primarily by blocking the glutamatergic NMDA receptor. It may also upregulate α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor throughput.85 Although glutamate is the major excitatory neurotransmitter, its role in anhedonia has yet to be fully explored. Preliminary evidence suggests that glutamate may have an integral role in both anhedonia and depression.47,50 Ketamine is also a partial agonist of the dopamine D2 receptor86,87 and has been found to increase dopamine levels in the striatum, including the caudate and the putamen.88 Intriguingly, Meyer et al.89 found that MDD patients with co-occurring motor retardation symptoms exhibited lower extracellular dopamine in the putamen compared with healthy volunteers. Taken together, these findings suggest that the glutamatergic system and its downstream modulation of dopaminergic activity may be one potential route of the anti-anhedonic efficacy of ketamine.
The present work has several limitations that should be noted. First, the lack of a baseline PET image meant that we could not exclude carryover effects in this crossover design. Second, the experiential difference between receiving ketamine and saline can be dramatic, and subjects likely realized what infusion they were receiving in each session, potentially invalidating the placebo arm of this study (however, see the recent article by Murrough et al.42). Third, all participants continued to receive either lithium or valproate, which may have masked or enhanced the effect of ketamine. It is unknown whether the rapid-acting anti-anhedonic effects of ketamine would also occur in unmedicated BD patients. Finally, we do not know how applicable these findings may be to MDD patients. Thus, we recommend that future studies attempting to replicate and extend the promising findings presented here incorporate additional PET or functional imaging scans to evaluate changes from baseline; study medication-free patients; and examine both MDD and BD patients. Furthermore, exploring the precise underlying reward processing improvements found here, for example, via cognitive testing, is essential to our ability to appropriately characterize the anti-anhedonic effects of ketamine.
Due to the prevalence of anhedonia in psychiatric and neurological illnesses, particularly in schizophrenia, Parkinson’s disease, drug addiction and both mood and anxiety disorders, anhedonia has been suggested as a tractable endophenotype. As outlined in the research domain criteria, identifying symptom-based etiologies and their treatment may help to clarify specific biological mechanisms mediating mental illnesses, aid to rectify classification and diagnostic issues currently inherent in psychiatry and provide sustainable stepping stones toward treatment improvements in mental health.
In sum, this study demonstrated that ketamine exerts rapid-acting anti-anhedonic effects in treatment-refractory BD patients. These anti-anhedonic effects remained even when the variance associated with depression score changes was removed, suggesting that ketamine ameliorates anhedonia independent of its already considerable antidepressant effects. Furthermore, these anti-anhedonic effects appeared to be mediated by increased glucose metabolism in the dACC and putamen. Our results underscore the putative utility of NMDA receptor antagonists to treat all facets of depression.
References
Mazza M, Squillacioti MR, Pecora RD, Janiri L, Bria P . Effect of aripiprazole on self-reported anhedonia in bipolar depressed patients. Psychiatry Res 2009; 165: 193–196.
Snaith RP . Identifying depression: the significance of anhedonia. Hosp Pract (Off Ed) 1993; 28: 55–60.
McMakin DL, Olino TM, Porta G, Dietz LJ, Emslie G, Clarke G et al. Anhedonia predicts poorer recovery among youth with selective serotonin reuptake inhibitor treatment-resistant depression. J Am Acad Child Adolesc Psychiatry 2012; 51: 404–411.
Spijker J, Bijl RV, de Graaf R, Nolen WA . Determinants of poor 1-year outcome of DSM-III-R major depression in the general population: results of the Netherlands Mental Health Survey and Incidence Study (NEMESIS). Acta Psychiatr Scand 2001; 103: 122–130.
Uher R, Perlis RH, Henigsberg N, Zobel A, Rietschel M, Mors O et al. Depression symptom dimensions as predictors of antidepressant treatment outcome: replicable evidence for interest-activity symptoms. Psychol Med 2012; 42: 967–980.
Nutt D, Demyttenaere K, Janka Z, Aarre T, Bourin M, Canonico PL et al. The other face of depression, reduced positive affect: the role of catecholamines in causation and cure. J Psychopharmacol 2007; 21: 461–471.
Opbroek A, Delgado PL, Laukes C, McGahuey C, Katsanis J, Moreno FA et al. Emotional blunting associated with SSRI-induced sexual dysfunction. Do SSRIs inhibit emotional responses? Int J Neuropsychopharmacol 2002; 5: 147–151.
Price J, Cole V, Goodwin GM . Emotional side-effects of selective serotonin reuptake inhibitors: qualitative study. Br J Psychiatry 2009; 195: 211–217.
McCabe C, Mishor Z, Cowen PJ, Harmer CJ . Diminished neural processing of aversive and rewarding stimuli during selective serotonin reuptake inhibitor treatment. Biol Psychiatry 2010; 67: 439–445.
Hindmarch I . The behavioural toxicity of antidepressants: effects on cognition and sexual function. Int Clin Psychopharmacol 1998; 13 (Suppl 6): S5–S8.
Hu XH, Bull SA, Hunkeler EM, Ming E, Lee JY, Fireman B et al. Incidence and duration of side effects and those rated as bothersome with selective serotonin reuptake inhibitor treatment for depression: patient report versus physician estimate. J Clin Psychiatry 2004; 65: 959–965.
Landen M, Hogberg P, Thase ME . Incidence of sexual side effects in refractory depression during treatment with citalopram or paroxetine. J Clin Psychiatry 2005; 66: 100–106.
Fawcett J, Scheftner WA, Fogg L, Clark DC, Young MA, Hedeker D et al. Time-related predictors of suicide in major affective disorder. Am J Psychiatry 1990; 147: 1189–1194.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. (5th edn), American Psychiatric Publishing: Arlington, VA, USA, 2013.
Der-Avakian A, Markou A . The neurobiology of anhedonia and other reward-related deficits. Trends Neurosci 2012; 35: 68–77.
Treadway MT, Zald DH . Reconsidering anhedonia in depression: lessons from translational neuroscience. Neurosci Biobehav Rev 2011; 35: 537–555.
Argyropoulos SV, Nutt DJ . Anhedonia revisited: Is there a role for dopamine-targeting drugs for depression? J Psychopharmacol 2013; 27: 869–877.
Berlin I, Givry-Steiner L, Lecrubier Y, Puech AJ . Measures of anhedonia and hedonic responses to sucrose in depressive and schizophrenic patients in comparison with healthy subjects. Eur Psychiatry 1998; 13: 303–309.
Dichter GS, Smoski MJ, Kampov-Polevoy AB, Gallop R, Garbutt JC . Unipolar depression does not moderate responses to the Sweet Taste Test. Depress Anxiety 2010; 27: 859–863.
Sherdell L, Waugh CE, Gotlib IH . Anticipatory pleasure predicts motivation for reward in major depression. J Abnorm Psychol 2012; 121: 51–60.
Etain B, Roy I, Henry C, Rousseva A, Schürhoff F, Leboyer M et al. No evidence for physical anhedonia as a candidate symptom or an endophenotype in bipolar affective disorder. Bipolar Disord 2007; 9: 706–712.
Wise RA . Dopamine, learning and motivation. Nat Rev Neurosci 2004; 5: 483–494.
Flagel SB, Clark JJ, Robinson TE, Mayo L, Czuj A, Willuhn I et al. A selective role for dopamine in stimulus-reward learning. Nature 2011; 469: 53–57.
Salamone JD, Correa M . The mysterious motivational functions of mesolimbic dopamine. Neuron 2012; 76: 470–485.
Schultz W, Dayan P, Montague PR . A neural substrate of prediction and reward. Science 1997; 275: 1593–1599.
Salamone JD, Cousins MS, Snyder BJ . Behavioral functions of nucleus accumbens dopamine: empirical and conceptual problems with the anhedonia hypothesis. Neurosci Biobehav Rev 1997; 21: 341–359.
Carter RM, Macinnes JJ, Huettel SA, Adcock RA . Activation in the VTA and nucleus accumbens increases in anticipation of both gains and losses. Front Behav Neurosci 2009; 3: 21.
O'Doherty JP, Deichmann R, Critchley HD, Dolan RJ . Neural responses during anticipation of a primary taste reward. Neuron 2002; 33: 815–826.
Schott BH, Minuzzi L, Krebs RM, Elmenhorst D, Lang M, Winz OH et al. Mesolimbic functional magnetic resonance imaging activations during reward anticipation correlate with reward-related ventral striatal dopamine release. J Neurosci 2008; 28: 14311–14319.
Knutson B, Adams CM, Fong GW, Hommer D . Anticipation of increasing monetary reward selectively recruits nucleus accumbens. J Neurosci 2001; 21: RC159.
Ernst M, Nelson EE, McClure EB, Monk CS, Munson S, Eshel N et al. Choice selection and reward anticipation: an fMRI study. Neuropsychologia 2004; 42: 1585–1597.
Dunlop BW, Nemeroff CB . The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry 2007; 64: 327–337.
Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL . Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depress Anxiety 2000; 11: 58–65.
Zarate CA Jr, Payne JL, Singh J, Quiroz JA, Luckenbaugh DA, Denicoff KD et al. Pramipexole for bipolar II depression: a placebo-controlled proof of concept study. Biol Psychiatry 2004; 56: 54–60.
Goldberg JF, Burdick KE, Endick CJ . Preliminary randomized, double-blind, placebo-controlled trial of pramipexole added to mood stabilizers for treatment-resistant bipolar depression. Am J Psychiatry 2004; 161: 564–566.
Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P . A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br J Psychiatry 1995; 167: 99–103.
Lemke MR, Brecht HM, Koester J, Reichmann H . Effects of the dopamine agonist pramipexole on depression, anhedonia and motor functioning in Parkinson's disease. J Neurol Sci 2006; 248: 266–270.
Boyer P, Tassin JP, Falissart B, Troy S . Sequential improvement of anxiety, depression and anhedonia with sertraline treatment in patients with major depression. J Clin Pharm Ther 2000; 25: 363–371.
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006; 63: 856–864.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 2000; 47: 351–354.
Ibrahim L, Diazgranados N, Franco-Chaves J, Brutsche N, Henter ID, Kronstein P et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology 2012; 37: 1526–1533.
Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry 2013; 170: 1134–1142.
Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, aan het Rot M et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry 2013; 74: 250–256.
Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry 2010; 67: 793–802.
Zarate CA Jr, Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, Cravchik A et al. Replication of ketamine's antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry 2012; 71: 939–946.
Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry 2010; 167: 748–751.
Yuksel C, Ongur D . Magnetic resonance spectroscopy studies of glutamate-related abnormalities in mood disorders. Biol Psychiatry 2010; 68: 785–794.
Paul IA, Skolnick P . Glutamate and depression: clinical and preclinical studies. Ann N Y Acad Sci 2003; 1003: 250–272.
Walter M, Henning A, Grimm S, Schulte RF, Beck J, Dydak U et al. The relationship between aberrant neuronal activation in the pregenual anterior cingulate, altered glutamatergic metabolism, and anhedonia in major depression. Arch Gen Psychiatry 2009; 66: 478–486.
Bechtholt-Gompf AJ, Walther HV, Adams MA, Carlezon WA Jr, Ongür D, Cohen BM . Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology 2010; 35: 2049–2059.
John CS, Smith KL, Van't Veer A, Gompf HS, Carlezon WA Jr, Cohen BM et al. Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacology 2012; 37: 2467–2475.
Garcia LS, Comim CM, Valvassori SS, Réus GZ, Stertz L, Kapczinski F et al. Ketamine treatment reverses behavioral and physiological alterations induced by chronic mild stress in rats. Prog Neuropsychopharmacol Biol Psychiatry 2009; 33: 450–455.
Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry 2011; 69: 754–761.
Papp M, Moryl E . New evidence for the antidepressant activity of MK-801, a non-competitive antagonist of NMDA receptors. Pol J Pharmacol 1993; 45: 549–553.
Reus GZ, Abelaira HM, Stringari RB, Fries GR, Kapczinski F, Quevedo J . Memantine treatment reverses anhedonia, normalizes corticosterone levels and increases BDNF levels in the prefrontal cortex induced by chronic mild stress in rats. Metab Brain Dis 2012; 27: 175–182.
Moghaddam B, Adams B, Verma A, Daly D . Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 1997; 17: 2921–2927.
Magistretti PJ, Pellerin L . Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci 1999; 354: 1155–1163.
Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF et al. Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc Natl Acad Sci USA 1999; 96: 8235–8240.
Nugent AC, Diazgranados N, Carlson PJ, Ibrahim L, Luckenbaugh DA, Brutsche N et al. Neural correlates of rapid antidepressant response to ketamine in bipolar disorder. Bipolar Disord 2013; 16: 119–128.
Montgomery SA, Åsberg M . A new depression scale designed to be sensitive to change. Br J Psychiatry 1979; 134: 382–389.
Sackeim HA . The definition and meaning of treatment-resistant depression. J Clin Psychiatry 2001; 62 (Suppl 16): 10–17.
First MB, Spitzer RL, Gibbon M, Williams JB . Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition. (SCID-I/P). Biometrics Research, New York State Psychiatric Institute: New York, NY, USA, 2002.
Franken IH, Rassin E, Muris P . The assessment of anhedonia in clinical and non-clinical populations: further validation of the Snaith-Hamilton Pleasure Scale (SHAPS). J Affect Disord 2007; 99: 83–89.
Leventhal AM, Chasson GS, Tapia E, Miller EK, Pettit JW . Measuring hedonic capacity in depression: a psychometric analysis of three anhedonia scales. J Clin Psychol 2006; 62: 1545–1558.
Nakonezny PA, Carmody TJ, Morris DW, Kurian BT, Trivedi MH . Psychometric evaluation of the Snaith-Hamilton pleasure scale in adult outpatients with major depressive disorder. Int Clin Psychopharmacol 2010; 25: 328–333.
Brooks RA . Alternative formula for glucose utilization using labeled deoxyglucose. J Nucl Med 1982; 23: 538–539.
Robinson OJ, Cools R, Carlisi CO, Sahakian BJ, Drevets WC . Ventral striatum response during reward and punishment reversal learning in unmedicated major depressive disorder. Am J Psychiatry 2012; 169: 152–159.
Pizzagalli DA, Holmes AJ, Dillon DG, Goetz EL, Birk JL, Bogdan R et al. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am J Psychiatry 2009; 166: 702–710.
Bremner JD, Vythilingam M, Vermetten E, Nazeer A, Adil J, Khan S et al. Reduced volume of orbitofrontal cortex in major depression. Biol Psychiatry 2002; 51: 273–279.
Drevets WC . Orbitofrontal cortex function and structure in depression. Ann N Y Acad Sci 2007; 1121: 499–527.
Luckenbaugh DA, Ibrahim L, Brutsche N, Franco-Chaves J, Mathews D, Marquardt CA et al. Family history of alcohol dependence and antidepressant response to an N-methyl-D-aspartate antagonist in bipolar depression. Bipolar Disord 2012; 14: 880–887.
Phelps LE, Brutsche N, Moral JR, Luckenbaugh DA, Manji HK, Zarate CA Jr . Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biol Psychiatry 2009; 65: 181–184.
Landen M, Nissbrandt H, Allgulander C, Sörvik K, Ysander C, Eriksson E . Placebo-controlled trial comparing intermittent and continuous paroxetine in premenstrual dysphoric disorder. Neuropsychopharmacology 2007; 32: 153–161.
Pecina S, Berridge KC . Hedonic hot spot in nucleus accumbens shell: where do mu-opioids cause increased hedonic impact of sweetness? J Neurosci 2005; 25: 11777–11786.
Shidara M, Richmond BJ . Anterior cingulate: single neuronal signals related to degree of reward expectancy. Science 2002; 296: 1709–1711.
Haruno M, Kawato M . Heterarchical reinforcement-learning model for integration of multiple cortico-striatal loops: fMRI examination in stimulus-action-reward association learning. Neural Netw 2006; 19: 1242–1254.
Haruno M, Kawato M . Different neural correlates of reward expectation and reward expectation error in the putamen and caudate nucleus during stimulus-action-reward association learning. J Neurophysiol 2006; 95: 948–959.
Kennerley SW, Walton ME, Behrens TE, Buckley MJ, Rushworth MF . Optimal decision making and the anterior cingulate cortex. Nat Neurosci 2006; 9: 940–947.
Benoit RG, Gilbert SJ, Burgess PW . A neural mechanism mediating the impact of episodic prospection on farsighted decisions. J Neurosci 2011; 31: 6771–6779.
Strunk DR, Lopez H, DeRubeis RJ . Depressive symptoms are associated with unrealistic negative predictions of future life events. Behav Res Ther 2006; 44: 861–882.
Thimm JC, Holte A, Brennen T, Wang CE . Hope and expectancies for future events in depression. Front Psychol 2013; 4: 470.
Keedwell PA, Andrew C, Williams SC, Brammer MJ, Phillips ML . The neural correlates of anhedonia in major depressive disorder. Biol Psychiatry 2005; 58: 843–853.
Fawcett J, Clark DC, Scheftner WA, Gibbons RD . Assessing anhedonia in psychiatric patients. Arch Gen Psychiatry 1983; 40: 79–84.
Liu RJ, Fuchikami M, Dwyer JM, Lepack AE, Duman RS, Aghajanian GK . GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology 2013; 38: 2268–2277.
Maeng S, Zarate CA Jr, Du J, Schloesser RJ, McCammon J, Chen G et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 2008; 63: 349–352.
Kapur S, Seeman P . Ketamine has equal affinity for NMDA receptors and the high-affinity state of the dopamine D2 receptor. Biol Psychiatry 2001; 49: 954–957.
Kapur S, Seeman P . NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Mol Psychiatry 2002; 7: 837–844.
Vollenweider FX, Vontobel P, Oye I, Hell D, Leenders KL . Effects of (S)-ketamine on striatal dopamine: a [11C]raclopride PET study of a model psychosis in humans. J Psychiatr Res 2000; 34: 35–43.
Meyer JH, McNeely HE, Sagrati S, Boovariwala A, Martin K, Verhoeff NP et al. Elevated putamen D(2) receptor binding potential in major depression with motor retardation: an [11C]raclopride positron emission tomography study. Am J Psychiatry 2006; 163: 1594–1602.
Acknowledgements
The authors thank Wayne Drevets MD and Maura Furey PhD for helpful discussions related to the manuscript, Ioline Henter, MA for outstanding editorial assistance and the 7SE research unit and staff for their support. Funding for this work was supported by the Intramural Research Program at the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH; grant number 04-M-0222), by a National Alliance for Research on Schizophrenia and Depression Award to CAZ, and by a Wellcome Trust-NIH PhD studentship (WT095465) to NL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A patent application for the use of ketamine in depression has been submitted listing CAZ among the inventors; he has assigned his rights on the patent to the US government, but will share a percentage of any royalties that may be received by the government. The remaining authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Lally, N., Nugent, A., Luckenbaugh, D. et al. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Transl Psychiatry 4, e469 (2014). https://doi.org/10.1038/tp.2014.105
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.105
This article is cited by
-
Plasticity of synapses and reward circuit function in the genesis and treatment of depression
Neuropsychopharmacology (2023)
-
Ketamine’s acute effects on negative brain states are mediated through distinct altered states of consciousness in humans
Nature Communications (2023)
-
Ventral prefrontal network response to alcohol in young adults with bipolar disorder: a within-subject randomized placebo-controlled alcohol administration study
Neuropsychopharmacology (2023)
-
A Pain Physician and Anesthesiologist’s Perspective on Ketamine for Treatment-Resistant Depression
SN Comprehensive Clinical Medicine (2023)
-
A translational perspective on the anti-anhedonic effect of ketamine and its neural underpinnings
Molecular Psychiatry (2022)
