
Abstract
Whole-genome expression profiling in postmortem brain tissue has recently provided insight into the pathophysiology of schizophrenia. Previous microarray and RNA-Seq studies identified several biological processes including synaptic function, mitochondrial function and immune/inflammation response as altered in the cortex of subjects with schizophrenia. Now using RNA-Seq data from the hippocampus, we have identified 144 differentially expressed genes in schizophrenia cases as compared with unaffected controls. Immune/inflammation response was the main biological process over-represented in these genes. The upregulation of several of these genes, IFITM1, IFITM2, IFITM3, APOL1 (Apolipoprotein L1), ADORA2A (adenosine receptor 2A), IGFBP4 and CD163 were validated in the schizophrenia subjects using data from the SNCID database and with quantitative RT-PCR. We identified a co-expression module associated with schizophrenia that includes the majority of differentially expressed genes related to immune/inflammation response as well as with the density of parvalbumin-containing neurons in the hippocampus. The results indicate that abnormal immune/inflammation response in the hippocampus may underlie the pathophysiology of schizophrenia and may be associated with abnormalities in the parvalbumin-containing neurons that lead to the cognitive deficits of the disease.
Similar content being viewed by others


Introduction
Schizophrenia is a common and devastating brain disease caused by the complex interplay of multiple genetic and environmental factors.1 Despite considerable research effort, the pathophysiology of the disease is not well understood. Genetic studies undertaken to identify the possible cause of the disease have yielded numerous candidate genes and genetic variations.2, 3 However, most candidates have not been well replicated or validated.4 The most consistent finding from the genetic studies is an association between schizophrenia and SNPs on chromosome 6p spanning the major histocompatibility complex region.2, 3 This result suggests that genetic diversity in the immune system may confer risk of schizophrenia.
Although genome-wide expression profiling using microarray technology has identified abnormalities in several biological processes including synaptic function and mitochondrial function,5, 6, 7, 8 abnormalities have also been detected in immune/inflammation response in the brain of schizophrenia cases,9, 10, 11, 12, 13 although there are inconsistencies between the various studies. However, recent advances in massively parallel sequencing methodology such as messenger RNA (mRNA) sequencing (RNA-Seq) can provide more accurate, sensitive and reliable gene expression data than microarrays.14, 15 Indeed, recent RNA-Seq studies have revealed an increase in inflammatory markers in the prefrontal cortex16 and a dysregulation of immune-related genes in the blood of individuals with schizophrenia.17, 18, 19 Whole-genome expression data such as these may be used to explore possible associations between abnormal gene expression and other markers of neuropathology in the brain of schizophrenia cases.20 These associations may reveal molecular mechanisms that underlie the pathophysiology of mental disorders and point to targets for medication development. Although a number of neuropathological abnormalities have been identified in the brain of individuals with schizophrenia,21, 22, 23 a reduction in the density of GABAergic neurons24 and of perineuronal oligodendrocytes25 appear to be among the most robust findings. Our previous study using a correlation analysis between genome-wide microarray expression data and the density of brain cells in the frontal cortex revealed multiple biological processes as abnormal in schizophrenia.20
Here we sequence mRNA from the hippocampus of individuals with schizophrenia and unaffected controls to identify the differentially expressed genes. We then performed a co-expression network analysis to examine the effects of confounding variables and to confirm which biological processes are altered in the hippocampus of the schizophrenia cases.
Materials and methods
Brain samples
RNA for the sequencing study was extracted from postmortem hippocampal sections from subjects in the Stanley Neuropathology Consortium (SNC).26 Additional RNA for qRT-PCR was extracted from the hippocampal sections from subjects in the Array Collection. The SNC contains 15 well-matched cases in each of four groups: schizophrenia, bipolar disorder, major depression and unaffected controls.26 The Array Collection is an independent tissue collection containing 35 cases in each of three groups: schizophrenia, bipolar disorder and unaffected controls. The groups from both tissue collections are matched for descriptive variables such as age, gender, race, postmortem interval, mRNA quality, brain pH and hemisphere. The collections have been used for numerous neuropathology studies, gene expression microarray studies and SNP association studies. For RNA-Seq, hippocampal RNA was obtained from 14 schizophrenia cases and 15 unaffected controls in the SNC. One schizophrenia case was excluded because of poor quality of RNA. For qRT-PCR validation, RNA was extracted from 20 randomly selected schizophrenia cases and 19 controls from the Array Collection and included with the samples from the SNC to gain adequate detection power. Descriptive variables are listed in the Table 1.
Library preparation and mRNA sequencing
Extracted total RNA was analysed using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Total RNA (1 μg) was subjected to two rounds of hybridization to oligo(dT) beads (Dynal, Carlsbad, CA, USA). The resulting mRNA was then used as a template for cDNA synthesis. The mRNA was randomly fragmented to between 200 and 700 bp by focused acoustic shearing (Covaris, Woburn, MA, USA) and converted to first strand cDNA using Superscript III (Invitrogen, Carlsbad, CA, USA), followed by second-strand cDNA synthesis using Escherichia coli DNA pol I (Invitrogen). The double stranded cDNA library was further processed by Illumina Genomic DNA Sample Prep kit (San Diego, CA, USA). It involved end repair using T4 DNA polymerase, Klenow DNA polymerase and T4 polynucleotide kinase followed by a single <A> base addition using Klenow 3′ to 5′ exo- polymerase, and was ligated with Illumina’s adaptor oligo mix using T4 DNA ligase. Adaptor-ligated library was size selected by electrophotetic separaton on a 2% agarose gel and excising the library smear at 500 bp. The library was PCR amplified for 18 cycles using phusion polymerase and purified by Qiaquick PCR Purification Kit (Qiagen, Carlsbad, CA, USA) and quantified by Quant-iT picogreen dsDNA Assay Kit (Invitrogen) following the manufacturer's protocol.
We then prepared Genome Analyser (GA) paired-end flow cell on the supplied Illumina cluster station and generated 50-bp paired-end sequence reads on the Illumina GA platform following the manufacturer’s protocol. Images taken during the sequencing reactions were processed in three stages by Illumina’s software: Firecrest performed image analysis, base-calling was done by Bustard and the sequence analysis was performed with Gerald.
Reads mapping and quantification of gene expression
All reads were mapped to UCSC H. sapiens reference genome (build hg18) using TopHat v2.0.0 with UCSC refFlat gene model annotation file on the –G parameter.27 We used the expected mean inner distance between mate paired-ends as –r parameter. TopHat calls Bowtie v0.12.7 to perform the alignment with no more than two mismatches. We used the pre-built index files of UCSC H. sapiens hg18, which are downloaded from the TopHat homepage (http://tophat.cbcb.umd.edu/index.html). The quantification of gene expression was accomplished by HTseq v0.5.3p9 and edger package (http://www.bioconductor.org/packages/2.11/bioc/html/edgeR.html). All mapped read counts of the genes were counted by htseq-count (subprogram of HTseq) with UCSC refFlat gene model annotation file, no strand-specific option, and intersection-nonempty option.
Validation RNA-Seq results using Stanley Neuropathology Consortium Integrative Database
The Stanley Neuropathology Consortium Integrative Database (SNCID) (http://sncid.stanleyresearch.org/) is an online archival database with data mining tools for analysing neuropathology markers, microarray data and SNP data from the SNC.28 qRT-PCR data of APOL1 from frontal cortex (BA9 and BA11) and of IFITM2 from frontal cortex, occipital cortex and thalamus were analysed using non-parametric variance analysis tool in the SNCID. Density of parvalbumin (PV)-containing neurons in the hippocampus (five sub-regions) was also analysed using the analysis tool.
qRT-PCR validation
Amplified transcriptome was used for qRT-PCR validation because only limited amounts of RNA were available from the hippocampus of the same subjects. 20 ng of hippocampal RNA from each subject was diluted in DEPC treated water to 10 ng μl−1 and adjusted to a volume of 5 μl with nuclease-free water for each whole-transcriptome amplification. A standard 2 h reaction with the QuantiTect Whole Transcriptome Kit (Qiagen) allowed for a uniform amplification of all transcripts within each RNA sample. Quantification of the amplified cDNA was carried out as described in the whole transcriptome kit’s protocol using the Quanti-iT PicoGreen dsDNA reagent (Invitrogen). The samples were then diluted to 2 ng μl−1 in nuclease-free water for qPCR with SYBR Select Master Mix (Carlsbad, CA, USA) (ABI). Each sample was run four times in 20 μl qPCR reactions (SYBR Select 2X, 0.6 μM Primer, 10 ng cDNA) and loaded onto a 384 well plate using an epMotion 5075 (Eppendorf, Hauppauge, NY, USA). Fluorescence detection and qPCR were carried out in an ABI Prism 7900HT Sequence Detection System (ABI). After excluding potential outliers from each sample, we retained at least three technical replicates from each sample for analysis. qPCR was carried out in this method for seven genes of interest (ADORA2A, APOL1, CD163, IGFBP4, IFITM1, IFITM2, IFITM3). The specific primers for the genes were designed using Primer3 29 except for IFITM1 and IFITM3. PCR primers for IFITM1 were obtained from Qiagen (Hs_IFITM1_2_SG, QuantiTect primer assay). IFITM3 primers were designed based on a previous study30. PCR primers are listed in Supplementary Table 1. The data was normalized using three housekeeping genes (ACTB2, HPRT1, TFRC1) that were acquired from Qiagen’s bioinformatically verified collection of QuantiTect Primer assays. qPCR reactions were run on 2% agarose gels (100 ml 0.5 × TAE, 2 g agarose, 5 μl EtBr) and the dissociation curves were also checked for evidence of nonspecific amplification. For further validation samples were sequenced using the Sanger method. One schizophrenia case was excluded from the statistical analysis because no amplification was detected for most of the genes.
Statistical analysis
Ct values from qRT-PCR were converted to the linear values using standard curves. Expression levels of ADORA2A, APOL1, CD163, IGFBP4, IFITM1, IFITM2 and IFITM3 were normalized to geometric means of three housekeeping genes. The normalized expression levels were log 10 transformed because those were not normally distributed. Then, descriptive variables were tested for identification of potential confounding factors. The effects of the continuous variables such as age, brain pH, postmortem interval, body mass index, lifetime exposure to antipsychotics and RNA integrity number (RIN) on the transformed qRT-PCR data were examined by Pearson correlation test. The categorical variable, sex and smoking were tested by t-test. The qRT-PCR data was statistically analysed with an analysis of covariance model (StatView, SAS, Cary, NC, USA). Differences in the gene expression levels between schizophrenia and controls were examined using an analysis of covariance model (StatView). P-values less than 0.05 were considered significant.
Gene co-expression network analysis
Unsupervised co-expression network analysis was performed using the Weighted Correlation Network Analysis in R.31 The co-expression network was generated using normalized RNA-Seq data of all genes in the hippocampus of schizophrenia and normal controls. The minimum module size and the minimum height for merging modules were set at 30 and 0.25, respectively. Correlation between co-expression modules and traits such as disease, demographic variables and clinical variables were performed to identify modules associated with schizophrenia and/or confounding factors. Raw data for the density of PV-containing neurons in the hippocampus were used as a trait after downloading from the SNCID in order to identify modules that may be associated with the density of these neurons. The co-expression module was visualized using VisANT.32
Functional annotation and enrichment map
DAVID (http://david.abcc.ncifcrf.gov/home.jsp) was used to identify the biological processes that were significantly over-represented by differentially expressed genes between schizophrenia and normal controls as well as genes included in the co-expression modules.33 False discovery rates less than 0.1 were considered significant. To visualize biological processes and to overcome gene-set redundancy, the enrichment map was then generated with the DAVID output file.34 Enrichment significance threshold was set at P-value less than 0.005 and false discovery rate less than 0.25. Overlap coefficient was set at 0.5
Immunohistochemistry
Fresh frozen sections of the hippocampus from two control and two schizophrenia cases were stained with antibodies to identify the specific cell types that express the protein encoded by six of the differentially expressed genes. The six selected were S100A8, S100A9, CD163, HP (haptoglobin), APOL1 (Apolipoprotein L1) and ADORA2A (adenosine receptor 2A). Details of the antibodies used and dilutions are described in Supplementary Table 2. Sections were thawed and fixed in 4% paraformaldehyde before primary antibody was applied overnight at 4 °C. Following washes, sections were incubated in biotinylated secondary antibody, washed and incubated with avidin–biotin–peroxidase complex (Vectastain ABC kit, Vector Laboratories, Burlingame, CA, USA), treated with 3,3′-Diaminobenzidine, washed, stained with Nissl and coverslipped.
Results
Differential gene expression in the hippocampus of individuals with schizophrenia
We performed whole-genome expression profiling using RNA-Seq data from the hippocampus of individuals with schizophrenia and unaffected controls to identify differentially expressed genes. For each sample, an average of 62.6 million 53-bp reads was generated (Supplementary Table 3). Sequence reads were mapped to the human reference genome (hg18) and for each sample, the average number of mapped reads was 58.1 million. An average of 89% of all the reads that were mapped was uniquely mapped. We then quantified the aligned read counts and performed statistical analysis. A total of 144 genes were significantly differentially expressed between schizophrenia and unaffected controls in the hippocampus at FDR<0.05 (Supplementary Table 4). The expression of 21 genes was downregulated, whereas 123 genes were upregulated in the hippocampus of individuals with schizophrenia as compared with unaffected controls (Supplementary Table 4). A functional annotation was performed to identify biological processes, which were significantly enriched in the differentially expressed genes. The enrichment map was generated to visualize functional clusters of the enriched biological processes. Three clusters of biological processes were identified: circulatory system process, defence response and polysaccharide catabolic process (Figure 1). Defence response/inflammatory response/immune response was the main cluster in the enrichment map and the genes involved in the process were predominantly upregulated in the hippocampus of schizophrenic patients as compared with unaffected controls (Figure 1).

(a) Biological processes over-represented in differentially expressed genes in the hippocampus between schizophrenia cases and controls and (b) heat map of differentially expressed genes related to defence response. Enrichment map for differentially expressed genes in the hippocampus between schizophrenia and controls and the heat map were generated with the Enrichment Map Plugin34 for Cytoscape programme.81 Nodes represent functional groups of gene sets and edge thickness is proportion to the overlap between gene sets in the enrichment map.
Validation of RNA-Seq results using SNCID database and qRT-PCR
We then validated the differentially expressed genes using data from the SNCID database28 and qRT-PCR. The SNCID is an archival and mining database, which includes numerous neuropathology data from the same individuals.28 Hence, the data can be used for independent validation for the RNA-Seq results. The RNA-Seq data showed that PV mRNA expression was significantly reduced in the hippocampus of the schizophrenia subjects as compared with the controls. The density of PV-containing GABAergic neurons were also significantly reduced in the hippocampus of the same individuals with schizophrenia as compared with the same unaffected controls, which is consistent with the RNA-Seq results (All P<0.05, except hippocampus/subiculum) (Table 2). Data for two other genes that were differentially expressed in the RNA-Seq analysis were also available in the database, although the data was derived from other brain regions. APOL1 mRNA expression was significantly upregulated in the frontal cortex (BA9) of schizophrenia (P=0.02) (Table 2) and IFITM2 mRNA was significantly increased in the thalamus of schizophrenia cases as compared with controls (P=0.002) (Table 2).
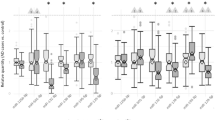
We validated the differential expression of seven genes from our RNA-Seq results using qRT-PCR. To gain adequate detection power, we included additional samples from the Array Collection for the qRT-PCR validation. Confounding effects of demographic and clinical variables were examined. RIN was significantly correlated with expression levels of the seven genes and brain pH was additionally correlated with APOL1 (All P<0.05). Body mass index was correlated with expression level of ADORA2A and smoking was associated with expression level of CD163 and IFITM3. Analysis of covariance revealed that ADORA2A, APOL1, IGFBP4, IFITM1, IFITM2 and IFITM3 mRNA levels were significantly increased in schizophrenia (Figure 2, All P<0.05), which is consistent with the RNA-Seq results. Expression of CD163 mRNA was increased in schizophrenia at trend level (Figures 2, P=0.09).

Quantitative real-time (qRT)-PCR validation of RNA-Seq results. Expression levels of selective. Genes related to immune/inflammation were measured by quantitative RT-PCR. #P<0.1, *P<0.05, **P<0.005, ***P<0.0001.
Co-expression network analysis
To identify altered biological processes in the hippocampus of schizophrenia, we conducted unsupervised gene co-expression network analyses using the normalized RNA-Seq data from both the schizophrenia and control groups. A total of 23 co-expression modules were generated and five modules were significantly associated with schizophrenia (Supplementary Table 5). However, three modules were excluded from downstream analysis because they were also significantly correlated with RIN. Module 3 (M3) was a large module consisting of 632 genes (Figure 3a). Ubiquitination, chromatin modification and protein localization were significantly enriched in the genes of this module (Figure 3b and Supplementary Figure 1). M19 was also a large module and included mainly the differentially expressed genes (Figure 4a). The I-kappa B/NF-kappa B cascade, cell-mediated immune response and inflammation were significantly enriched in the module (Figure 4b and Supplementary Figure 2). Immune-related genes such as S100A8, S100A9, IFITM2 and IFITM3 were included in the module.

Co-expression network analysis in the hippocampus. The co-expression module (M3), which is significantly associated with schizophrenia in the hippocampus (a) and major biological processes (Gene ontology) over-represented in the genes in the co-expression module (b). The top 150 network connections with topological overlap above the threshold of 0.03 were visualized using VisANT32. Genes related to chromatin/histone modification are blue.

Co-expression network analysis in the hippocampus. The co-expression module (M19), which is significantly associated with schizophrenia in the hippocampus (a) and major biological processes (Gene ontology) over-represented in the genes in the co-expression module (b). The top 150 network connections with topological overlap above the threshold of 0.03 were visualized using VisANT.32 Differentially expressed genes are blue.
Co-expression modules associated with reduced density of PV-containing neurons
Gene expression profiling can be used to explore the molecular mechanisms that may underlie neuropathological abnormalities. Thus, we performed a correlation analyses between the co-expression modules, which were associated with schizophrenia, M3 and M19, and the density of PV-containing neurons in five subfields of the hippocampus in the same individuals from which the RNA-Seq data was derived. The density of PV-containing neurons in the five subfields of the hippocampus was significantly reduced in schizophrenia as compared with the normal controls (Supplementary Table 6). Both M3 and M19 were significantly associated with the density of PV-containing neurons in CA1 and M19 was also correlated with the density of PV-containing neurons in CA3. This suggests that processes associated with these modules; transcriptional regulation, epigenetic control, immune response and inflammation, may be molecular mechanisms that contribute to the abnormalities found in the PV-containing neurons in the hippocampus of schizophrenia.
Immunohistochemistry of hippocampal tissues
Contrary to expectations, the immune/inflammation-related proteins were not expressed in microglia in the representative cases chosen for this survey. S100A8- and S100A9-positively labelled cells were found in the lumen of blood vessels (Supplementary Figure 3a, c) and in capillaries (Supplementary Figure 3b, d). The stain could also extend into the parenchyma that surrounded some of the vessels that contained labelled cells. CD163 labelled perivascular monocyte-like cells and both CD163 and HP positively labelled the endothelial cells of blood vessels, but not all blood vessels (Supplementary Figure 3e, f). HP stain could also extend into the parenchyma surrounding some of the positively labelled vessels. APOL1 and ADORA2A positively labelled perivascular astrocytic processes (Supplementary Figure 3g) and astrocytes adjacent to pial and ependymal surfaces (Supplementary Figure 3h).
Discussion
Genome-wide gene expression profiling is an unbiased approach to identify the pathophysiology underlying complex diseases. In this study, we identified genes related to immune response and inflammation that are upregulated in the hippocampus of schizophrenia as compared with unaffected controls. Moreover, our co-expression network analysis identified gene expression modules enriched for the same biological processes that were associated with the variable ‘schizophrenia’ and for the variable ‘density of PV-containing neurons’. The RNA-Seq methodology for reliable gene expression profiling has several advantages over microarrays because it is a more sensitive technique and has less confounding effects on gene expression levels. The method has no hybridization bias and almost no batch effects on gene expression.14 15
Previous postmortem studies using microarray9, 10, 11, 12, 13 and RNA-Seq16 identified immune/inflammation-related genes that were upregulated in the cerebral cortex of schizophrenia subjects. Our current study replicates these findings in the hippocampus of individuals with schizophrenia. The activation of immune/inflammation-related genes implicates possible environmental insults due to infection or other stressors in the aetiology of schizophrenia. Exposure to viruses or protozoan has been implicated in the aetiology of psychiatric disorders35, 36 and numerous studies have found an increase in peripheral markers of immune activation in schizophrenia.37, 38, 39 The relative risk of developing schizophrenia also increases after prenatal infection40 and animal models of maternal immune activation result in developmental abnormalities in the brain structure and function that reflect those found in psychiatric disorders.41, 42 As we find the immune signal appears to be coming from cells in the blood and at the blood–brain barrier (BBB), the changes we find in the hippocampus could reflect a chronic systemic infection, where infectious agents may increase the risk of schizophrenia by activating immune/inflammation-related markers in the blood and at the BBB that then interact with genetic susceptibility and cause downstream effects on neurons and glia. Alternatively, the changes may result from an immune challenge that occurred during development that then disrupts brain development and leaves an abnormal immune signature in the genetically susceptible individuals.43, 44 Additional factors such as stress or autoimmune responses could also impact immune/inflammation-related genes, perhaps synergistically to produce neuropathology in genetically susceptible individuals.45
We identified a number of cell types expressing the proteins encoded by the differentially expressed immune/inflammation-related genes in the hippocampus of individuals with schizophrenia. Contrary to previous studies16, 46, 47 that have suggested that the microglia are likely to be responsible for the increased expression of immune/inflammation-related genes in the brain of schizophrenia cases, we do not find any of the proteins expressed in microglia, at least not in the representative samples chosen here. S100A8 and S100A9 are calcium-binding proteins known to have antimicrobial activity and to be expressed in myeloid cells, particularly macrophages.48, 49 We show that both proteins appear to be localized to blood monocyte-like cells in the brain, although in some instances they also appear to be excreted into the surrounding brain parenchyma. Monocytes are significantly increased in the blood and cerebrospinal fluid in schizophrenia.50, 51, 52 Another upregulated gene that we find expressed in these same monocyte-like cells is Chitinase-3-like-1 (CHI3L1; data not shown). CHI3L1 is upregulated in response to a number of environmental stresses including inflammation and hypoxia53, 54 and a recent meta-analysis shows that polymorphisms in CHI3L1 confer risk for schizophrenia.55 Similarly, plasma levels of HP are elevated in schizophrenia and are associated with polymorphisms in the HP gene.56, 57 HP, which we find expressed in the endothelial cells of the brain, is a blood plasma protein involved in iron homoeostasis and inflammatory disease58 and binds free haemoglobin to inhibit oxidative activity and prevent tissue damage.59 Interestingly, clearance of the haemoglobin–HP complex is mediated by the macrophage scavenger receptor, CD163, which is upregulated in a range of inflammatory diseases.60 CD163 is expressed on perivascular macrophages that are strategically positioned between the endothelial cells and the basement membranes of cerebral blood vessels61, 62 and appears to be expressed in the endothelial cells of some blood vessels as well. The IFITM genes that we find upregulated in the hippocampus are also known to be expressed in the vascular endothelial cells of the brain.44, 63 The IFITM1, 2 and 3 proteins are particularly interesting because they are known to defend against, and to be strongly induced by a number of viruses, including Influenza A and West Nile.64, 65, 66, 67 Moreover, immune challenge in neonatal mice causes an increase in IFITM3 protein, which appears to be essential for the resulting neuropathological impairments and brain dysfunction, as the impairments do not occur in ifitm3−/− mice receiving the same treatment.68
In addition to blood cells and endothelial cells, we also find a number of the upregulated markers expressed in the perivascular astrocytes. Apolipoprotein L1 (APOL1) is involved in lipid homoeostasis but circulating plasma APOL1 is also a trypanolytic factor that can kill the trypanosome pathogen that causes sleeping sickness.69 Interestingly, in the brain we find the APOL1 protein expressed in perivascular astrocytes and in astrocytes adjacent to the pial and ependymal surfaces. Similarly, ADORA2A, which is a G-protein coupled receptor subtype for adenosine, is also expressed in the perivascular astrocytes in the hippocampus. This is in contrast to its abundant distribution in the neurons of the basal ganglia.70 ADORA2A is also known to be expressed in lymphocytes and has been implicated in inflammatory disorders.71 Thus, in the hippocampus these markers, which have all been implicated in immune/inflammatory responses, are expressed in either blood cells or in the endothelial cells and perivascular astrocytes that are situated to monitor factors in the blood. The majority of differentially expressed immune/inflammation-related genes that we find are expressed at this BBB but are only expressed in a small percentage of cells in any one section. The endothelial cells, for example account for only 0.1% of all brain cells72 and not all endothelial cells were positively labelled, which underscores the sensitivity of the RNA-seq technology that is able to detect the differential expression of these genes. Our findings corroborate a previous microdissection study that focused on vascular endothelial cells of the frontal cortex and detected abnormal expression of inflammatory response genes in the schizophrenia cases as compared with controls73 and also an electron microscope study, which found utrastructural abnormalities in endothelial cells and astrocytic endfeet in the frontal cortex of patients with schizophrenia.74 A genetic–inflammatory–vascular theory of schizophrenia has been proposed previously75 and posits that damage to the microvasculature in the brain may result from genetically influenced abnormal inflammatory responses that are triggered by environmental stressors such as infection, trauma or hypoxia. Our results add support to this theory, as we find cells within, and surrounding, the vasculature that are directly implicated in the dysregulation of the immune/inflammation-related genes. Moreover, these findings indicate that abnormalities previously described in the other cells types of the brain, particularly in the GABAergic neurons in schizophrenia, may be downstream of these BBB abnormalities.
PV-containing GABAergic neurons contribute to the gamma oscillations that are necessary for normal cognitive processing.76 The abnormalities of PV-containing GABAergic interneurons found in various brain regions of individuals with schizophrenia may contribute to some of the cognitive deficits found in the disease.77, 78 A previous postmortem study found a significant decrease in the density of PV-containing neurons in multiple sub-regions of the hippocampus of the same individuals from which we sequenced the RNA.79 When we examined the correlation between the density of PV-containing neurons and the co-expression modules associated with schizophrenia, we found two co-expression modules that were significantly associated with the disease status as well as with the density of PV-containing neurons in the hippocampus. The module M19 included genes associated with immune response and/or inflammation response including S100A8, S100A9, IFITM2 and IFITM3. Both S100A8 and S100A9 appear to be labelling blood monocyte-like cells whereas IFITM2 and IFITM3 are expressed in endothelial cells in the brain,44, 63 which suggests that these cells are upregulating the markers in response to something in their environment that then effects the function of the PV-containing neurons in the hippocampus and consequently may be related to the cognitive deficits associated with schizophrenia. Preliminary data from the prefrontal cortex has also shown that there is an inverse correlation between the higher IFITM mRNA levels in the endothelial cells and markers of the GABAergic neurons in schizophrenia.63 Interestingly, a recent animal study of mice that have a mild chronic inflammation of the brain (Schnurri-2; an NF-kappa B site-binding protein knockout), have no microglial activation but a decrease in PV levels and multiple cognitive deficits.80
The results of our RNA-Seq study, which are consistent with previous gene expression profiling studies,8, 9, 10, 13, 16 have shown upregulation of immune/inflammation-related genes in schizophrenia and although there may be limitations to the study due to potential confounding effects on the RNA-Seq data, we believe we have managed to overcome them by performing a system level analysis using the co-expression network analysis to examine the potential confounding effects on the gene expression data. Among the five modules that were correlated with schizophrenia, three modules, M6, M11 and M13 were also correlated with RIN. As RIN is a technical confounding variable, we excluded these three modules from further analysis. The M19 module, which included immune/inflammation-related genes, was also correlated with brain pH. Thus the results may be interpreted with caution and further studies with additional cohorts and brain regions may be required to exclude the possible confounding effects on gene expression profiling. The hippocampus tissue used for the RNA-Seq study included multiple cell types so a future cell-type-specific RNA-Seq study employing laser capture microdissection will provide increased sensitivity and specificity.
In conclusion, this RNA-Seq study supports a hypothesis that activated immune/inflammation may underlie the pathophysiology of schizophrenia and contribute to the cognitive deficits of the disease. It appears that much of the immune/inflammation signal is coming from cells within and adjacent to the cerebral vasculature. Determining if there is a primary abnormality in the cells of the BBB or whether they are responding to signals in the periphery will require further study. Medications that target the immune/inflammatory system may be important for the future prevention or treatment of schizophrenia.
References
Mueser KT, McGurk SR . Schizophrenia. Lancet 2004; 363: 2063–2072.
Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D et al. Common variants conferring risk of schizophrenia. Nature 2009; 460: 744–747.
Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet 2008; 40: 827–834.
Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P . Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 2000; 28: 53–67.
Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL et al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry 2004; 9: 684–697, 643.
Iwamoto K, Bundo M, Kato T . Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet 2005; 14: 241–253.
Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P . Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci 2002; 22: 2718–2729.
Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E . Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry 2007; 7: 46.
Arion D, Unger T, Lewis DA, Levitt P, Mirnics K . Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry 2007; 62: 711–721.
Shao L, Vawter MP . Shared gene expression alterations in schizophrenia and bipolar disorder. Biol Psychiatry 2008; 64: 89–97.
Schmitt A, Leonardi-Essmann F, Durrenberger PF, Parlapani E, Schneider-Axmann T, Spanagel R et al. Regulation of immune-modulatory genes in left superior temporal cortex of schizophrenia patients: a genome-wide microarray study. World J Biol Psychiatry 2011; 12: 201–215.
Arion D, Horvath S, Lewis DA, Mirnics K . Infragranular gene expression disturbances in the prefrontal cortex in schizophrenia: signature of altered neural development? Neurobiol Dis 2010; 37: 738–746.
Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y . RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 2008; 18: 1509–1517.
Wang Z, Gerstein M, Snyder M . RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009; 10: 57–63.
Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry 2013; 18: 206–214.
Xu J, Sun J, Chen J, Wang L, Li A, Helm M et al. RNA-Seq analysis implicates dysregulation of the immune system in schizophrenia. BMC Genomics 2012; 13 (Suppl 8): S2.
Sainz J, Mata I, Barrera J, Perez-Iglesias R, Varela I, Arranz MJ et al. Inflammatory and immune response genes have significantly altered expression in schizophrenia. Mol Psychiatry 2012; 18: 1056–1057.
Gardiner EJ, Cairns MJ, Liu B, Beveridge NJ, Carr V, Kelly B et al. Gene expression analysis reveals schizophrenia-associated dysregulation of immune pathways in peripheral blood mononuclear cells. J Psychiatr Res 2013; 47: 425–437.
Kim S, Webster MJ . Correlation analysis between genome-wide expression profiles and cytoarchitectural abnormalities in the prefrontal cortex of psychiatric disorders. Mol Psychiatry 2010; 15: 326–336.
Knable MB, Barci BM, Bartko JJ, Webster MJ, Torrey EF . Molecular abnormalities in the major psychiatric illnesses: Classification and Regression Tree (CRT) analysis of post-mortem prefrontal markers. Mol Psychiatry 2002; 7: 392–404.
Knable MB, Barci BM, Webster MJ, Meador-Woodruff J, Torrey EF . Molecular abnormalities of the hippocampus in severe psychiatric illness: postmortem findings from the Stanley Neuropathology Consortium. Mol Psychiatry 2004; 9: 609–620, 544.
Knable MB, Torrey EF, Webster MJ, Bartko JJ . Multivariate analysis of prefrontal cortical data from the Stanley Foundation Neuropathology Consortium. Brain Res Bull 2001; 55: 651–659.
Beasley CL, Zhang ZJ, Patten I, Reynolds GP . Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry 2002; 52: 708–715.
Vostrikov VM, Uranova NA, Orlovskaya DD . Deficit of perineuronal oligodendrocytes in the prefrontal cortex in schizophrenia and mood disorders. Schizophr Res 2007; 94: 273–280.
Torrey EF, Webster M, Knable M, Johnston N, Yolken RH . The stanley foundation brain collection and neuropathology consortium. Schizophr Res 2000; 44: 151–155.
Trapnell C, Pachter L, Salzberg SL . TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009; 25: 1105–1111.
Kim S, Webster MJ . The Stanley neuropathology consortium integrative database: a novel, web-based tool for exploring neuropathological markers in psychiatric disorders and the biological processes associated with abnormalities of those markers. Neuropsychopharmacology 2010; 35: 473–482.
Rozen S, Skaletsky H . Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
Li D, Peng Z, Tang H, Wei P, Kong X, Yan D et al. KLF4-mediated negative regulation of IFITM3 expression plays a critical role in colon cancer pathogenesis. Clin Cancer Res 2011; 17: 3558–3568.
Langfelder P, Horvath S . WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559.
Hu Z, Mellor J, Wu J, DeLisi C . VisANT: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics 2004; 5: 17.
Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC et al. Database for annotation, visualization, and integrated discovery. Genome Biol 2003; 4: P3.
Merico D, Isserlin R, Stueker O, Emili A, Bader GD . Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 2010; 5: e13984.
Torrey EF, Miller J, Rawlings R, Yolken RH . Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr Res 1997; 28: 1–38.
Brown AS, Derkits EJ . Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 2010; 167: 261–280.
Drexhage RC, van der Heul-Nieuwenhuijsen L, Padmos RC, van Beveren N, Cohen D, Versnel MA et al. Inflammatory gene expression in monocytes of patients with schizophrenia: overlap and difference with bipolar disorder. A study in naturalistically treated patients. Int J Neuropsychopharmacol 2010; 13: 1369–1381.
Kurian SM, Le-Niculescu H, Patel SD, Bertram D, Davis J, Dike C et al. Identification of blood biomarkers for psychosis using convergent functional genomics. Mol Psychiatry 2011; 16: 37–58.
Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B . Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 2011; 70: 663–671.
Cannon M, Clarke MC . Risk for schizophrenia--broadening the concepts, pushing back the boundaries. Schizophr Res 2005; 79: 5–13.
Shi L, Fatemi SH, Sidwell RW, Patterson PH . Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci 2003; 23: 297–302.
Kneeland RE, Fatemi SH . Viral infection, inflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2013; 42: 35–48.
Michel M, Schmidt MJ, Mirnics K . Immune system gene dysregulation in autism and schizophrenia. Dev Neurobiol 2012; 72: 1277–1287.
Horvath S, Mirnics K . Immune system disturbances in schizophrenia. Biol Psychiatry 2013 doi:10.1016/j.biopsych.2013.06.010(e-pub ahead of print).
Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 2013; 339: 1095–1099.
Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC . Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med 2009; 50: 1801–1807.
Monji A, Kato TA, Mizoguchi Y, Horikawa H, Seki Y, Kasai M et al. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry 2013; 42: 115–121.
Lagasse E, Clerc RG . Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol Cell Biol 1988; 8: 2402–2410.
Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 2007; 13: 1042–1049.
Zorrilla EP, Cannon TD, Gur RE, Kessler J . Leukocytes and organ-nonspecific autoantibodies in schizophrenics and their siblings: markers of vulnerability or disease? Biol Psychiatry 1996; 40: 825–833.
Nikkila HV, Muller K, Ahokas A, Miettinen K, Rimon R, Andersson LC . Accumulation of macrophages in the CSF of schizophrenic patients during acute psychotic episodes. Am J Psychiatry 1999; 156: 1725–1729.
Drexhage RC, Hoogenboezem TA, Cohen D, Versnel MA, Nolen WA, van Beveren NJ et al. An activated set point of T-cell and monocyte inflammatory networks in recent-onset schizophrenia patients involves both pro- and anti-inflammatory forces. Int J Neuropsychopharmacol 2011; 14: 746–755.
Junker N, Johansen JS, Hansen LT, Lund EL, Kristjansen PE . Regulation of YKL-40 expression during genotoxic or microenvironmental stress in human glioblastoma cells. Cancer Sci 2005; 96: 183–190.
Recklies AD, Ling H, White C, Bernier SM . Inflammatory cytokines induce production of CHI3L1 by articular chondrocytes. J Biol Chem 2005; 280: 41213–41221.
Ohi K, Hashimoto R, Yasuda Y, Yoshida T, Takahashi H, Iike N et al. The chitinase 3-like 1 gene and schizophrenia: evidence from a multi-center case-control study and meta-analysis. Schizophr Res 2010; 116: 126–132.
Maes M, Delanghe J, Bocchio Chiavetto L, Bignotti S, Tura GB, Pioli R et al. Haptoglobin polymorphism and schizophrenia: genetic variation on chromosome 16. Psychiatry Res 2001; 104: 1–9.
Wan C, La Y, Zhu H, Yang Y, Jiang L, Chen Y et al. Abnormal changes of plasma acute phase proteins in schizophrenia and the relation between schizophrenia and haptoglobin (Hp) gene. Amino Acids 2007; 32: 101–108.
Wassell J . Haptoglobin: function and polymorphism. Clin Lab 2000; 46: 547–552.
Asleh R, Marsh S, Shilkrut M, Binah O, Guetta J, Lejbkowicz F et al. Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ Res 2003; 92: 1193–1200.
Moestrup SK, Moller HJ . CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann Med 2004; 36: 347–354.
Lau SK, Chu PG, Weiss LM . CD163: a specific marker of macrophages in paraffin-embedded tissue samples. Am J Clin Pathol 2004; 122: 794–801.
Fabriek BO, Van Haastert ES, Galea I, Polfliet MM, Dopp ED, Van Den Heuvel MM et al. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia 2005; 51: 297–305.
Volk DW, Siegel BI, Sengupta EJ, Edelson JR, Lewis DA . Elevated transcript levels for viral restriction factors in cortical endothelial cells in schizophrenia. Neuropsychopharmacology 2012; 38: T32.
Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009; 139: 1243–1254.
Feeley EM, Sims JS, John SP, Chin CR, Pertel T, Chen LM et al. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog 2011; 7: e1002337.
Bailey CC, Huang IC, Kam C, Farzan M . Ifitm3 limits the severity of acute influenza in mice. PLoS Pathog 2012; 8: e1002909.
Diamond MS, Farzan M . The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 2013; 13: 46–57.
Ibi D, Nagai T, Nakajima A, Mizoguchi H, Kawase T, Tsuboi D et al. Astroglial IFITM3 mediates neuronal impairments following neonatal immune challenge in mice. Glia 2013; 61: 679–693.
Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homble F et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 2005; 309: 469–472.
Schiffmann SN, Fisone G, Moresco R, Cunha RA, Ferre S . Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol 2007; 83: 277–292.
Varani K, Padovan M, Vincenzi F, Targa M, Trotta F, Govoni M et al. A2A and A3 adenosine receptor expression in rheumatoid arthritis: upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res Ther 2011; 13: R197.
Shusta EV, Boado RJ, Mathern GW, Pardridge WM . Vascular genomics of the human brain. J Cereb Blood Flow Metab 2002; 22: 245–252.
Harris LW, Wayland M, Lan M, Ryan M, Giger T, Lockstone H et al. The cerebral microvasculature in schizophrenia: a laser capture microdissection study. PLoS One 2008; 3: e3964.
Uranova NA, Zimina IS, Vikhreva OV, Krukov NO, Rachmanova VI, Orlovskaya DD . Ultrastructural damage of capillaries in the neocortex in schizophrenia. World J Biol Psychiatry 2010; 11: 567–578.
Hanson DR, Gottesman II . Theories of schizophrenia: a genetic-inflammatory-vascular synthesis. BMC Med Genet 2005; 6: 7.
Vreugdenhil M, Jefferys JG, Celio MR, Schwaller B . Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J Neurophysiol 2003; 89: 1414–1422.
Sohal VS, Zhang F, Yizhar O, Deisseroth K . Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009; 459: 698–702.
Volman V, Behrens MM, Sejnowski TJ . Downregulation of parvalbumin at cortical GABA synapses reduces network gamma oscillatory activity. J Neurosci 2011; 31: 18137–18148.
Zhang ZJ, Reynolds GP . A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res 2002; 55: 1–10.
Takao K, Kobayashi K, Hagihara H, Ohira K, Shoji H, Hattori S et al. Deficiency of Schnurri-2, an MHC enhancer binding protein, induces mild chronic inflammation in the brain and confers molecular, neuronal, and behavioral phenotypes related to schizophrenia. Neuropsychopharmacology 2013; 38: 1409–1425.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13: 2498–2504.
Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB et al. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 2003; 362: 798–805.
Acknowledgements
This study was supported by Stanley Medical Research Institute. We specially thank Drs. E. Fuller Torrey, Robert Yolken and Sarven Sabunciyan for helpful comments on study design and interpretation of results. We also thank Jonathan Cohen and Suad Diglisic for technical support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Hwang, Y., Kim, J., Shin, JY. et al. Gene expression profiling by mRNA sequencing reveals increased expression of immune/inflammation-related genes in the hippocampus of individuals with schizophrenia. Transl Psychiatry 3, e321 (2013). https://doi.org/10.1038/tp.2013.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2013.94
Keywords
This article is cited by
-
Schizophrenia endothelial cells exhibit higher permeability and altered angiogenesis patterns in patient-derived organoids
Translational Psychiatry (2024)
-
Significance of chitinase-3-like protein 1 in the pathogenesis of inflammatory diseases and cancer
Experimental & Molecular Medicine (2024)
-
Overexpression of transmembrane TNFα in brain endothelial cells induces schizophrenia-relevant behaviors
Molecular Psychiatry (2023)
-
Brain microvascular endothelial cells and blood-brain barrier dysfunction in psychotic disorders
Molecular Psychiatry (2023)
-
Brain Inflammatory Marker Abnormalities in Major Psychiatric Diseases: a Systematic Review of Postmortem Brain Studies
Molecular Neurobiology (2023)
