
Abstract
Adult neurogenesis has been implicated in affective disorders and the action of antidepressants (ADs) although the functional significance of this association is still unclear. The use of animal models closely mimicking human comorbid affective and anxiety disorders seen in the majority of patients should provide relevant novel information. Here, we used a unique genetic mouse model displaying higher trait anxiety (HAB) and comorbid depression-like behavior. We demonstrate that HABs have a lower rate of hippocampal neurogenesis and impaired functional integration of newly born neurons as compared with their normal anxiety/depression-like behavior (NAB) controls. In HABs, chronic treatment with the AD fluoxetine alleviated their higher depression-like behavior and protected them from relapse for 3 but not 7 weeks after discontinuation of the treatment without affecting neurogenesis. Similar to what has been observed in depressed patients, fluoxetine treatment induced anxiogenic-like effects during the early treatment phase in NABs along with a reduction in neurogenesis. On the other hand, treatment with AD drugs with a particularly strong anxiolytic component, namely the neurokinin-1-receptor-antagonist L-822 429 or tianeptine, increased the reduced rate of neurogenesis in HABs up to NAB levels. In addition, challenge-induced hypoactivation of dentate gyrus (DG) neurons in HABs was normalized by all three drugs. Overall, these data suggest that AD-like effects in a psychopathological mouse model are commonly associated with modulation of DG hypoactivity but not neurogenesis, suggesting normalization of hippocampal hypoactivity as a neurobiological marker indicating successful remission. Finally, rather than to higher depression-related behavior, neurogenesis seems to be linked to pathological anxiety.
Similar content being viewed by others


Introduction
Depression and anxiety are the most common psychiatric disorders in modern society and share a high degree of comorbidity (up to 80–90%, for review, see Klenk et al.1). Antidepressants (ADs) are not effective in 30–40% of patients,2, 3 and only 33% of patients achieve remission in the first line of treatment with widely used selective serotonin reuptake inhibitors (SSRIs).4 Mood-elevating effects of ADs are observed mainly in depressed patients but are almost absent in non-depressed volunteers,5, 6, 7 suggesting that the neurobiological targets of ADs in pathophysiologically deranged systems might differ from those in intact systems.8 Therefore, it is important to study AD-mediated changes in the brains of animal models that are as close to the clinical situation as possible, that is, in terms of exhibiting depression and comorbid anxiety. One of those animal models displaying higher trait anxiety- and depression-like behavior consists of HAB mice and HAB rats that have been selectively bred (for review, see Landgraf et al.,9 Sartori et al.10 and Singewald11). Although chronic treatment with the SSRI paroxetine has been previously shown to reduce the enhanced depression-related behavior displayed by HAB rats,12 the question of whether the higher anxiety/depression-related behavior of adult HAB mice can be normalized pharmacologically has not been studied so far. As prolonged use of ADs is associated with possible adverse effects,13 discontinuation of AD treatment is attempted after remission of depressive symptoms has occurred; this, however, is associated with the risk of relapse.14 Relevant animal models in this area that has up to now been under-studied may provide clues on markers, which may help in the decision as to whether to discontinue treatment without risking relapse or to continue treatment to protect from relapse.15, 16, 17 Our first aim was therefore to examine whether the aberrant anxiety/depression-related behavior in HABs can be reversed by ADs of different pharmacological classes, and to discover how long the behavioral effects last before relapse is observed. Specifically, both fluoxetine, an SSRI, and tianeptine, which seems to predominantly affect synaptic plasticity via the modulation of glutamatergic neurotransmission apart from other actions including selective serotonin reuptake enhancement,18 were chosen for their established AD efficacy in patients with depression,19, 20 whereas the selective neurokinin-1 receptor antagonist L-822,429 has demonstrated AD and anxiolytic effects in rodents including HAB mice21 (for review, see Ebner et al.22) as well as in sub-populations of patients with depression and/or anxiety disorders.22, 23
Various lines of research have implicated the hippocampus in the pathogenesis of depressive disorders because (i) this brain region is highly stress sensitive and stress is an important pathophysiological factor in both depression and anxiety (for review, see Lupien et al.24); (ii) it is one of the key regions identified as a component of a network that is dysregulated in major depressive disorder (MDD);25, 26, 27 and (iii) it has been shown to have a smaller volume in patients with MDD.28 Although a direct association between AD treatment and modulation of hippocampal volume has not been observed,28 there is growing evidence that depression might be associated with reduced hippocampal neurogenesis.29, 30, 31, 32 It has been suggested that ADs might exert their efficacy via modulating neurogenesis in ‘normal’ rodents,33, 34, 35, 36 in psychopathological animal models of higher depression-like behavior37 and also in nonhuman primates.38 However, most of the studies have shown that increasing or decreasing neurogenesis using knockouts or X-ray or pharmacological treatment is not sufficient to lead to the development of a depression-like phenotype in animals36, 39, 40, 41, 42, 43, 44, 45 (for review, see Petrik et al.46 and Sahay and Hen47). As MDD and anxiety are comorbid in humans, the neurogenesis hypothesis may be particularly relevant in cases exhibiting a combination of affective and anxiety disorders.46, 48 Therefore, our second aim was to assess whether the higher depression/anxiety-related behavior of HABs as well as its pharmacological rescue is associated with modulated hippocampal neurogenesis.
Newly born cells in the dentate gyrus (DG) mature into functional neurons, which display activity-dependent responsiveness on specific challenges that can activate the hippocampus;49 we therefore hypothesized that a permanent disturbance in the replenishment of newly born hippocampal neurons should result in reduced excitability in this brain region. Indeed, hypoactivity, which was normalized by behaviorally successful fluoxetine treatment, has been observed in depressed patients50 (for review, see Stone et al.51). Therefore, the third aim was to determine whether newly born and/or pre-existing neurons in the DG of HABs would display altered functional activation, which then would be normalized in parallel to behavioral normalization by successful drug treatment. If so, altered DG activity could serve as a potential neurobiological biomarker for higher anxiety and for higher depression, and also for their behavioral rescue.
Materials and methods
Animals
Experiments were carried out on adult female HAB and normal anxiety/depression-like behavior (NAB) mice (11–14 weeks of age) as anxiety and depressive disorders have been observed more frequently in females than males in humans52 and at least in part in available rodent models.53, 54 HABs and NABs were selectively bred for their innate anxiety-related behavior at the Department of Pharmacology and Toxicology, University of Innsbruck, Innsbruck, Austria (for details, see Sartori et al.21). Animals were housed (up to four animals per cage) in a temperature (22±1 °C) and humidity (60%) controlled room under a 12-h light–dark cycle with lights on at 0700 hours. They had access to food and water ad libitum. The experiments that were conducted were approved by the local Ethical Committee on Animal Care and Use (Bundesministerium für Wissenschaft and Kultur, Austria) and were in compliance with international laws and policies and European Council Directive 86/609/EEC.
Drug treatment
Bromodeoxyuridine, BrdU (Sigma, Steinheim, Germany) was injected at a dose of 200 mg kg−1 (intraperitoneally) twice per day.55, 56 The SSRI fluoxetine (18 mg kg day−1, Sigma), the selective neurokinin-1 receptor-antagonist (L-822,429) (2 S,3 S)–N-{[2-cyclopropoxy–5-(5-trifluoromethyl)-tetrazol-1-yl]benzyl-2-phenylpiperidin-3-amine-dihydrochloride] (30 mg kg−1 day−1)57 and tianeptine (50 mg kg−1 day−1, kind gift from Stablon, Servier, Austria, GmbH)58 were administered via the drinking water. A drug intake of the dose indicated above equivalent to a drug measure was achieved by adapting the concentrations of the drug in the drinking solutions according to mean drinking volume and body weight per cage.59 It is possible that small variations in the individual dose that was obtained because of variations in individual water consumption. Therefore, body weight was assessed and doses adjusted accordingly throughout the course of the experiment to exclude large variations. Mice were kept under the assigned experimental condition (based on drug treatment) until completion of all experiments. Control mice received tap water.
Behavioral testing
The forced swim test (FST) and the light–dark test were selected for assessing the level of depression- and anxiety-related behavior, respectively, and they were conducted between 0830 and 1700 hours according to previous protocols in the lab59, 60 (further details are given in the Supplementary methods section).
Sucrose preference test
The sucrose preference test was conducted based on the modification of a previously published study.61 During this test, mice were given a free choice between two bottles, one with 1% sucrose and another with tap water. The position of the bottles were switched every 48 h to prevent the possible effects of side preference in drinking behavior. No previous food or water deprivation was applied before the test. The consumption of water and sucrose solution was estimated simultaneously in control and experimental groups by weighing the bottles. Each session denotes an average of sucrose preference with the bottle each time on the left and the right side. The preference for sucrose was calculated as a percentage of consumed sucrose solution of the total amount of liquid drank.
Immunohistochemistry
Two hours after the onset of the FST or light–dark test, animals were deeply anesthetized with an overdose of sodium pentobarbital (200 mg kg−1) and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde in 0.1 mol l−1 phosphate-buffered solution (pH= 7.4). Free-floating coronal sections (50 μm) were processed for BrdU, doublecortin (DCX) and c-Fos immunohistochemistry according to previous protocols.12, 62 They were incubated in one of the following primary antibodies: rat anti-BrdU (1/500, Harlan Sera-Lab, Loughborough, UK), goat anti-DCX C18 (1/250, Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-c-Fos (1/10 000, Santa Cruz), mouse anti-NeuN (1/500, Chemicon, Temecula, CA, USA). Subsequently, they were incubated in a corresponding biotinylated goat anti-rat secondary antibody (1/200, Vector Laboratories, Burlingame, CA, USA), rabbit anti-goat secondary antibody (1/200, Vector Laboratories) and goat anti-rabbit secondary antibody (1/200, Vector Laboratories). These antigen–antibody complexes were visualized by the avidin–biotin–horseradish peroxidase procedure (Vectastain Elite ABC-kit, Vector Laboratories) with 3,3’-diaminobenzidine as the chromogen. Alternatively, for double-/triple-labeling studies, primary antibodies were detected using Alexa-Fluor-conjugated secondary antibodies (Invitrogen, Molecular Probes, Karlsruhe, Germany) or Rhodamine donkey anti-rat, Cy2 donkey anti-goat and Cy5 donkey anti-mouse antibodies and sections were mounted in Prolong Gold (Invitrogen).
Quantification of cells
One-in-eight of the sections obtained of each brain were 3,3′-diaminobenzidine stained, and immunoreactive cells were stereologically counted using a computer-assisted image analysis system (Nikon E-800 microscope, CCD video camera, Optronics MicroFire, Goleta, CA, USA; Stereo Investigator Software, MicroBrightField Europe eK, Magdeburg, Germany) throughout the rostrocaudal extent on the dorsal and ventral blade of the granular layer of the DG,63, 64, 65 applying a 20 × and 40 × objective. A cell was considered as c-Fos-labeled (c-Fos positive) or as BrdU labeled, if the brown-black 3,3′-diaminobenzidine-stained nucleus was unambiguously darker than the background staining, and this included all cells from low to high intensities of staining. For immunofluoresence, imaging was performed with a Zeiss fluorescence microscope (AxioObserver, Vienna, Austria), attached to an ApoTome slider for structured illumination. The resulting images were analyzed with the Zeiss AxioVision software (version 4.8.1) and colocalization of BrdU+ and c-Fos+ nuclei was detected by visual inspection of the two overlapping fluorescent channels. BrdU/DCX/NeuN labeling was analyzed with a confocal laser scanning microscope (SP5 Laser scanning microscope, Leica Microsysterms, Wetzlar, Germany), using an argon laser (514 nm) for the selective excitation of Cy2, DPSS laser (561 nm) for the excitation of Rhodamine and a HeNe laser (633 nm) for the excitation of Cy5. The sections were scanned in 1 μm optical sections using a 40 × objective.
Statistical analyses
All data are presented as mean ±s.e.m. and were all first tested for homoscedasticity using Levene's test (using Statistica software, Statsoft Inc., OK, USA). As parametric distributions were revealed for all data, behavioral parameters and cell counts were further statistically analyzed by using an unpaired t-test, a repeated-measures analysis of variance or a two-way analysis of variance with line and treatment as dependent factors. A Duncan’s post hoc comparisons test was applied whenever possible. Correlation analysis was conducted using Pearson’s product moment. Statistical significance was set at P<0.05.
Results
HABs displayed higher depression-like behavior, a lower rate of newly born cell survival as well as neurogenesis and functional integration of newly born neurons, along with a lower challenge-induced DG activity
To analyze depression-like behavior in HAB and NAB mice, animals were subjected to a FST 21 days after the administration of BrdU (Figure 1a). HABs displayed a higher immobility time compared with NABs (Figure 1b) indicating higher depression-related behavior. In addition, HABs also showed less preference to sucrose reflecting anhedonia (Supplementary Figure 1). The higher depression-like behavior as well as anhedonia coincided with a lower number of BrdU+ cells in the DG of HABs compared with NABs indicating a lower rate of surviving newly born cells (Figure 1c, Figures 2a and b). Similarly, a lower number of DCX+ cells were observed in HABs, indicating a lower number of immature neurons (Figure 1d and Figures 2c and d). Triple-labeling using BrdU, DCX and NeuN to identify newly born mature neurons further confirmed the observation that indeed HABs have a lower rate of neurogenesis in comparison with NABs (Figure 1e and Figures 2e–g).

HABs, in comparison with NABs, showed higher depression-like behavior, reduced neurogenesis/functional integration of newly born neurons and reduced DG activation on exposure to the FST. (a) Schematic representation of the experimental design. HABs display (b) higher immobility time, (c) a lower number of BrdU+ cells, (d) a lower number of DCX+ cells, (e) a lower proportion of BrdU+ cells in DCX+/NeuN+ cells, (f) a lower number of c-Fos+ cells and (g) a lower proportion of BrdU+ cells colabeled with c-Fos+ cells. **P<0.01, ***P<0.001 HAB vs NAB, n=6–9 mice per group. Data are represented as mean±s.e.m. BrdU, bromodeoxyuridine; DCX, doublecortin; FST, forced swim test; HAB, high anxiety/depression-like behavior; NAB, normal anxiety/depression-like behavior.

Representative image showing (a, b) BrdU+ cells in NABs and HABs as represented by brownish spots in the subgranular and granular layer of the DG indicated by black arrows. (c, d) DCX+ cells in NABs and HABs as represented by brownish spots indicating the cell body in the subgranular layer (indicated by black arrows) and the dendrites projecting into the GCL and ML (indicated by white arrows) of the DG. (e) A majority of BrdU+ cells (indicated by white arrows) are also DCX+ suggesting that these cells have become immature neurons (f) magnified image of an immature neuron indicated by white arrow. (g) A BrdU+ cell colabeled with both DCX and NeuN showing that it has become a fully mature neuron. This figure also shows a decline in DCX expression as the newly born cell starts expressing NeuN. Scale bar in (b, d): 50 μm; (e–g): 10 μm. BrdU, bromodeoxyuridine; DCX, doublecortin; DG, dentate gyrus; GCL, granular cell layer; HAB, high anxiety/depression-like behavior; ML, molecular layer; NAB, normal anxiety/depression-like behavior.
After exposure to the FST, a lower number of c-Fos+ cells was observed in the DG of HABs compared with NABs, indicating that HABs display challenge-induced hypoactivation of neurons in the DG (Figure 1f).
By double-labeling BrdU+/c-Fos+ cells, we were also able to test activation of newly born neurons on exposure to swim stress. As shown in Supplementary Figure 5b, 6-week-old newly born neurons were activated in NABs, but not HABs, in response to FST. We hypothesized that the integration time of 6 weeks might not be enough for the functional integration of newly born neurons. Therefore, we extended the time period to 10 weeks. However, we still did not observe any evidence of functionally activated newly born neurons in HABs (Supplementary Figure 5d). These data suggest that HABs lack or have largely reduced functional integration of newly born cells.
Chronic fluoxetine normalized higher depression-like behavior and DG hypoactivity, but not neurogenesis or anxiety-related behavior, in HABs
We next tested whether chronic fluoxetine administration would normalize the higher anxiety/depression-related behavior in HABs and whether this would be accompanied by a normalization of neurogenesis and DG activation, assessed by immediate early gene mapping. Fluoxetine was administered for 21 days, followed by either a light–dark test or FST (Figure 3a and Supplementary Figure 2a). Chronic fluoxetine treatment reduced the higher depression-related behavior in HABs without affecting FST behavior in NABs (Figure 3b), but failed to attenuate the higher anxiety-related behavior in HABs (Supplementary Figure 2c).

Effect of chronic fluoxetine treatment on depression-like behavior, neurogenesis and DG activity on exposure to FST in HABs and NABs. (a) Schematic representation of the experimental design. Chronic fluoxetine treatment (b) decreased the immobility time in HABs but not NABs (c) did not alter the number of BrdU+ cells in HABs but reduced it in NABs, (d) did not alter the proportion of BrdU+ cells in DCX+/NeuN+ in HABs, but reduced it in NABs, (e) increased the number of c-Fos+ cells in HABs but not in NABs. *P<0.05, **P<0.01, ***P<0.001 HAB-Ctl vs NAB-Ctl. $P<0.05, $$P<0.01 NAB-Ctl vs NAB-fluoxetine. #P<0.05 HAB-Ctl vs HAB-fluoxetine. n=6–9 mice per group. Data are represented as mean±s.e.m. BrdU, bromodeoxyuridine; Ctl, control; DCX, doublecortin; DG, dentate gyrus; FST, forced swim test; HAB, high anxiety/depression-like behavior; NAB, normal anxiety/depression-like behavior.
Although the number of surviving BrdU+ cells in HABs was unaltered, surprisingly, fluoxetine treatment reduced the BrdU+ cells in NABs (Figure 3c). BrdU+/DCX+/NeuN+ labeling confirmed that HABs did indeed display a lower rate of neurogenesis, which, however, was not normalized on fluoxetine administration (Figure 3d).
Moreover, and in agreement with our previous findings (Figure 1f), HABs had a lower number of c-Fos+ cells than NABs did. This hyporesponse was normalized after fluoxetine treatment (Figure 3e).
Duration of the effects of chronic fluoxetine treatment on depression-related behavior, DG activity and neurogenesis after drug discontinuation
We next investigated whether the AD-like effect of fluoxetine in HABs persisted even after cessation of fluoxetine administration, and if it did persist, for how long.
Fluoxetine was administered for 3 weeks, followed by a 3-week drug-free period. FST was conducted at the end of this period, on the 42nd day (Figure 4a). Here, untreated HABs displayed a higher immobility time than NABs. The attenuating effect of fluoxetine on depression-related behavior was observed only in HABs, and was preserved for 3 weeks after cessation of fluoxetine treatment (Figure 4b).

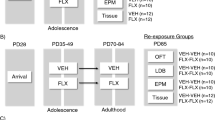
Effect of fluoxetine treatment for 3 weeks followed by discontinuation of the drug for either 3 or 7 weeks on depression-like behavior, neurogenesis and DG activity on exposure to FST in HABs and NABs. (a) Schematic representation of the experimental design for 3-week fluoxetine discontinuation period. A 3-week drug-free period (b) decreased the immobility time in HABs but not in NABs, (c) did not alter the number of BrdU+ cells in HABs but reduced it in NABs, (d) increased the number of c-Fos+ cells in HABs but not in NABs. (e) Depression-related behavior was negatively correlated with the number of c-Fos immunoreactive cells in the DG of HABs. (f) Schematic representation of the experimental design for the 7-week drug-free period. Chronic fluoxetine treatment for 3 weeks followed by a discontinuation for the next 7 weeks did not alter (g) the immobility time, (h) the number of BrdU+ cells or (i) or c-Fos+ cells. *P<0.05, **P<0.01, ***P<0.001 HAB-Ctl vs NAB-Ctl. $$P<0.01 NAB-Ctl vs NAB-fluoxetine. ##P<0.01 HAB-Ctl vs HAB-fluoxetine. n=6–9 mice per group. Data are represented as mean±s.e.m. BrdU, bromodeoxyuridine; Ctl, control; DG, dentate gyrus; FST, forced swim test; HAB, high anxiety/depression-like behavior; NAB, normal anxiety/depression-like behavior.
Analysis of neurogenesis in these animals revealed that, again, HABs showed a lower number of BrdU+ cells in comparison with NABs (Figure 4c). As also observed immediately after the 3-week fluoxetine treatment, the same treatment followed by a 3-week drug-free period had no effect on the number of BrdU+ cells in HABs but led to decrease in the number in NABs.
In addition, the animals were analyzed for the swim-stress-induced c-Fos response after the drug washout period. HABs showed a lower number of c-Fos+ cells than NABs, although this normalized and also remained normalized 3 weeks after cessation of fluoxetine treatment (Figure 4d). Furthermore, Pearsons’s product moment correlation revealed that there was a significant negative correlation between depression-related behavior and DG activity in HABs (Figure 4e). These results indicate that persistence of the AD-like effect of fluoxetine is accompanied by normalization of the challenge-induced neuronal hypoactivity in the DG.
Next, we increased the washout period that followed the 3-week fluoxetine treatment to 7 weeks (Figure 4f). Here, the AD effect was lost as there was no longer any detectable effect on the immobility time of HABs (Figure 4g). Furthermore, there was no effect on the rate of neurogenesis (Figure 4h) or on the c-Fos response in the DG of HABs (Figure 4i).
Effect of fluoxetine treatment during the early phase of treatment
One puzzling observation in our studies was that fluoxetine treatment reduced neurogenesis in NABs. As fluoxetine has been shown to exert an anxiogenic-like effect in the early phase of treatment in both humans66 and rodents67, 68 and as BrdU was administered during this phase of fluoxetine treatment (Figure 3a and Figure 4a), we hypothesized that the reduction in neurogenesis was associated with the early anxiogenic effects of fluoxetine. To test this hypothesis, we replicated the experimental set up (Figure 4a) with the modification that now BrdU was given on the 12th to the 16th day of fluoxetine administration (Figure 5a), when the ‘early’ anxiogenic- effect of fluoxetine had ceased.

Chronic fluoxetine treatment did not decrease neurogenesis in NABs when BrdU was administered 12–16 days after fluoxetine onset. (a) Schematic representation of the experimental design. Chronic fluoxetine treatment for 3 weeks (b) decreased the immobility time in HABs but not in NABs, (c) did not alter the number of BrdU+ cells in HABs or in NABs and (d) did not alter the number of DCX+ cells in HABs or NABs. **P<0.01, HAB-Ctl vs NAB-Ctl. ##P<0.01 HAB-Ctl vs HAB-fluoxetine. n=6-9 mice per group. Data are represented as mean±s.e.m. BrdU, bromodeoxyuridine; Ctl, control; DCX, doublecortin; FST, forced swim test; HAB, high anxiety/depression-like behavior; NAB, normal anxiety/depression-like behavior.
HABs again displayed higher immobility time than NABs. This was attenuated by fluoxetine (Figure 5b) even after a 3-week drug discontinuation period. In contrast to the observed decrease in neurogenesis in NABs when BrdU was given early, fluoxetine had no effect on cells that were labeled by BrdU between day 12 and day 16 (Figure 5c).
To further confirm the hypothesized early anxiogenic effect of fluoxetine on neurogenesis in NABs, fluoxetine was administered for 3 days and subsequently anxiety-like behavior was assessed in the light–dark test (supplementary Figure 3a). Fluoxetine was indeed shown to elicit an anxiogenic-like effect in NABs, indicated by a reduction in total time spent in the light compartment (Supplementary Figure 3c). There was no significant difference between lines in terms of the total distance traveled (Supplementary Figure 3b), demonstrating that general locomotor activity was not affected by fluoxetine treatment. In line with our hypothesis, the number of newly born cells and immature neurons decreased after this short-term fluoxetine administration (Supplementary Figures 3d and 3e).
Treatment with ADs with a particularly strong anxiolytic component normalized reduced neurogenesis and DG hypoactivation in HABs
Finally, we tested whether other ADs with a pharmacological mechanism that was different from SSRIs, namely the neurokinin-1-receptor antagonist L-822,429 and tianeptine would reduce the higher depression- and anxiety-related behavior and in contrast to fluoxetine they would indeed normalize the rate of neurogenesis in HABs.
Treatment with L-822,429 or tianeptine for 21 days (Figure 6a) led to an anxiolytic-like effect in HABs in the light–dark test (Figures 6b and c) without affecting locomotor activity as assessed in the open field test (supplementary Figure 4b and 4c). The anxiolytic effect of L-822,429 in HABs is in accordance with our previous findings.69 In addition, using separate set of experiments L-822,429 and tianeptine exerted an AD-like effect, namely, reduction of immobility time in the FST, selectively in HABs, without affecting this behavior in NABs (Figure 6d). L-822,429 as well as tianeptine increased neurogenesis in HABs, but not in NABs (Figure 6e). Also, both drugs profoundly increased the FST-induced c-Fos expression in the DG of HABs but not in NABs (Figure 6f).

Chronic treatment with L-822,429 or tianeptine had anxiolytic and antidepressant-like effects in HABs along with an increase in neurogenesis and normalization of challenge-induced DG hypoactivity. (a) Schematic representation of the experimental design. Chronic (b) L-822,429 or (c) tianeptine treatment increased the total time spent in the light compartment in the LD test in HABs, (d) reduced the immobility time in the FST in HABs without affecting NABs, (e) increased the proportion of BrdU+ cells in DCX+/NeuN+ cells in HABs without affecting NABs and (f) increased the number of c-Fos+ cells in the DG of HABs but not of NABs. *P<0.05, **P<0.01, HAB-Ctl vs NAB-Ctl. #P<0.05, ##P<0.01, ###P<0.001, HAB-Ctl vs HAB- L-822,429/ HAB-tia. n=6–9 mice per group. Data are represented as mean±s.e.m. BrdU, bromodeoxyuridine; Ctl, control; DCX, doublecortin; DG, dentate gyrus; FST, forced swim test; IHC, immunohistochemistry; LD, light–dark; HAB, high anxiety/depression-like behavior; NAB, normal anxiety/depression-like behavior; Tia, tianeptine.
Discussion
In the last decade, neurogenesis has been shown to be involved in the pathogenesis of anxiety and depression-related behavior under stressful conditions32 and/or the therapeutic action of ADs34 (for review, see Petrik et al.46 and Sahay and Hen47). We now extend these previous reports in an animal model of trait anxiety and comorbid depression and demonstrate the association of anxiety, but not of depression-related behavior, with neurogenesis in psychopathological, rather than basal physiological, conditions. The rescue of higher depression-related behavior through application of ADs of different pharmacological classes is observed. Interestingly, the AD-like effect is present for a few weeks after discontinuation of fluoxetine treatment before HABs relapse to the depressive-like phenotype. In addition, higher depression-related behavior seems to be associated with dysregulation of the DG under emotional challenge, and the AD-like effect is closely associated with successful normalization of the DG hypoactivation, but not with neurogenesis, in both the remission and the relapse phase.
Chronic AD treatment normalizes the higher depression-like behavior of HABs
In this study, we demonstrated that HABs display an increased behavioral despair in the FST and decreased sucrose preference, a measure of anhedonia. Anhedonia is a hallmark symptom of depression in humans and observed as decreased preference for sucrose solution compared over water in rodents.61, 70 Both these measures indicate that HABs have a higher depression-like behavior in comparison with their normal anxiety NAB counterparts. As different forced swim behaviors between HAB and NAB mice were not observed in early stages of selective breeding,71, 72 it is most likely due to an additional divergence in behavior in sequential generations of selective breeding, and closely reflects the high degree of comorbidity in patients suffering from anxiety and depression. This finding underscores the validity of using HABs in studies related to anxiety/depression disorders. In addition, we report sensitivity of depression-like behavior in HABs, but not in NABs, to chronic treatment with different classes of established (fluoxetine and tianeptine) and novel (L-822,429) ADs suggesting that in this model system, neuropathology that is related to the disorder must be present for the AD effects to occur.73 Similarly, ADs do not lead to significant mood-changing effects in non-depressed healthy individuals.5, 6, 7 Along these lines, it was suggested that paradigms elaborated in ‘non-depressed’ mice quite likely engage different neurobiological mechanisms that might be less relevant to clinical remission36 further supporting the validity of the HAB/NAB model.
Duration of AD-like and neurobiological effects after AD drug discontinuation
Fluoxetine is one of the most widely prescribed AD in clinical settings74 and has been experimentally widely used for studying the link between depression and neurogenesis (for review, see Benninghoff et al.75). We therefore selected this drug to further investigate whether and for how long AD-like effects persist after drug discontinuation in HABs, and if so, for how long. Although some work has been done in terms of studying remission/relapse in depressed patients after discontinuation of pharmacotherapy (for review, see Palazidou15), this subject needs to be studied more extensively in animals. For example, it has been shown that an AD-like effect still persists in rats 2 days after discontinuation of desipramine or venlafaxine treatment;76, 77 this acute phase of drug discontinuation, however, does not closely reflect remission in humans, which is usually observed weeks after drug discontinuation.16 On the basis of half-lives of fluoxetine and its active metabolite norfluoxetine (in mice, 8 h and 16 h, respectively),78 we chose time periods of 3 and 7 weeks after discontinuation of treatment ensuring almost complete elimination of both fluoxetine and norfluoxetine from plasma in HABs by this time. An AD effect in HABs was observed 3 weeks, but not 7 weeks, after fluoxetine discontinuation suggesting a loss of central, long-term adaptive changes responsible for the AD effect of fluoxetine over time.
In searching for possible neurobiological markers, that would indicate the switch from remission to relapse, we found a reversal of the challenge-induced hypoactivation of DG neurons in HABs to be such a candidate. Specifically, although the remission phase of fluoxetine was accompanied by a normalization of the hypoactive DG in HABs, this normalization was lost after switching from remission to relapse. In addition, reversal of DG hypoactivation was common to all ADs and paralleled that of the depressive phenotype in HABs. In a similar way, a hypoactive DG in stressed rodents79, 80 has been shown to be normalized by agomelatine treatment.79 Notably, the finding of a hypoactive DG in HAB mice following swim stress is in accordance with our earlier study, in which we reported hypoactivity in the DG on the introduction of the mild emotional stimulus of open arm exposure, but not under the basal/resting state.12 Together, these two studies point toward an aberrant hippocampal regulation in HABs during stressful conditions. Along the same lines, imaging data in MDD patients showed reduced hippocampal activation under challenge conditions,81, 82 but not during the resting state (for review, see Kuhn and Gallinat83). Indeed, as mentioned in the introduction, the hippocampus, via its connection with various corticolimbic structures, modulates the response to emotion, stress and motivation.25, 26 Taken together, these findings clearly indicate that changes in the challenge-induced activity patterns of DG neurons might provide an important neurobiological marker for depression-like behavior and AD effects.
AD-like effects in relation to adult neurogenesis and neuronal integration
We revealed that the higher depression-like behavior in HAB mice was accompanied by a lower rate of neurogenesis. This is in agreement with the earlier findings in HAB rats that showed a lower rate of neurogenesis31 demonstrating that this inverse relationship is valid in different species. Furthermore, these results are in line with the ‘neurogenic-depression hypothesis’.84, 85 Among the mechanisms underlying reduced neurogenesis (for review, see Zhao et al.86), brain-derived neurotrophic factor seems to be one promising candidate, as its lower levels have been found to be associated with depression and reduced neurogenesis87 (for review, see Pittenger and Duman88). Interestingly, hippocampal brain-derived neurotrophic factor expression is reduced in HAB in comparison with NAB mice (Sartori et al, unpublished), further substantiating this hypothesis.
Extending these findings, we have now shown for the first time that there was no functional activation of newly born neurons in the DG of HABs while those of NABs could be activated on a challenge. This finding raises the speculation that the time period of 6 weeks might not have been a sufficiently long period for functional integration of newly born neurons in HAB mice to occur, although this effect has been previously demonstrated from 2 to 3 weeks onward in different mouse strains including the CD-1 strain.55, 89, 90 On increasing the time period to 10 weeks, we still did not find any evidence of functional integration of newly born neurons in the DG of HABs. In an attempt to further substantiate these findings, we subjected HABs to a water maze test that is known to activate such neurons.55, 89 Again, we did not reveal any evidence of functional integration in HABs (data not shown). Therefore, it appears that the functional integration of newly born neurons is impaired or largely reduced in HABs. On the other hand, fluoxetine treatment did not affect the functional neuronal integration in either HABs or NABs. Two interesting observations deduced from our study are (1) fluoxetine treatment rescued the higher depression-like behavior in HABs, without increasing neurogenesis and (2) fluoxetine did not have any effect on NABs in the FST, but decreased neurogenesis to the level seen in HABs. Both these observations indicate a clear dissociation between modulation of neurogenesis and depression-like behavior. Thus, the hypothesis that hippocampal neurogenesis is required to exert the behavioral effects of fluoxetine is not supported by the data obtained in our psychopathological mouse model. In addition, we found a clear dissociation between modulation of neurogenesis and behavior, with fluoxetine treatment rescuing the depressive phenotype of HABs without increasing neurogenesis, but, conversely, fluoxetine not having any effect on NABs in the FST, but decreasing neurogenesis. Thus, the hypothesis that hippocampal neurogenesis is required to exert the behavioral effects of fluoxetine is not supported by the data obtained in our psychopathological mouse model. Future studies will reveal whether (i) HABs have an increased apoptosis rate and/or whether (ii) apoptosis is enhanced in NABs by early anxiogenic effects of fluoxetine administration.
Human studies investigating the involvement of neurogenesis in depression and the action of ADs have yielded mixed results. Work on post-mortem tissue found no change in proliferation in MDD patients,91 and recent data indicate that AD treatments enhance discrete facets of neurogenesis in human92 and non-human primate models.38, 92, 93 However, caution is urged in the interpretation of these due to low sample size, lack of information on agonal index or serum toxicology at death, and variations in the region of the hippocampus chosen for analysis.94 Thus, further studies are still needed before a firm conclusion can be drawn on the relationship between depression and neurogenesis in humans.94
Neurogenesis might be linked to anxiety-related behavior
Several of our observations in a psychopathological animal model suggest that neurogenesis is related to anxiety rather than depression-like behavior. In particular, only those drugs that elicited an anxiolytic effect in combination with the AD effect, namely, L-822,429 and tianeptine, also increased neurogenesis in HABs while fluoxetine, exerting an AD effect only, failed to increase neurogenesis in HABs and even decreased it in NABs. The latter effect raised speculations about a possible association with early anxiogenic effects of fluoxetine observed in rodents67, 68 and humans.66 To test this idea, we adopted two approaches. First, fluoxetine was given only for a short period along with BrdU, and second BrdU was administered on the 12th to the 16th day of chronic fluoxetine administration, when the anxiogenic effect was lost. Neurogenesis (as revealed by both BrdU and DCX labeling) was indeed decreased during the early phase of fluoxetine administration, and this was accompanied by an anxiogenic-like effect in NABs; and this decrease was reversed when BrdU was administered at a later phase reverting to a state in which the anxiogenic effects of fluoxetine were no longer detected in NABs. It is important to note here that both BrdU and DCX cells show a decline following 3 days of fluoxetine treatment. According to the study by Brown et al.,95 BrdU+ cells show 80–90% colocalization with DCX right after 3–4 days of BrdU injection. Since a decrease in BrdU+ cells following subchronic fluoxetine has been observed during this time period it could also indicate that nearly all the DCX+ cells, which were colocalized with BrdU also declined. This partially explains a rapid effect of fluoxetine on the reduced number of immature neurons. Furthermore, other studies have also indicated an apoptotic role of fluoxetine in the brains of healthy rodents.96, 97, 98 In contrast to this study, another study has shown that co-treatment with diazepam prevents the effect of fluoxetine on the proliferation and survival of hippocampal neurons.99 The author suggests that diazepam produces sedative effects that could indirectly impair neurogenesis by decreasing locomotor activity. Moreover, the earlier study99 has used C57Bl/6 mice, which have the highest proliferation rate,100 while this study has been conducted on CD1 mice, which have the highest survival rate.100 These differences might explain the contradiction between the present data in comparison with the earlier study.99 Altogether, these data indicate an association between the anxiogenic-like effect of fluoxetine and a reduction in neurogenesis. To further confirm this hypothesis, we treated HABs with L-822,429 and tianeptine, which have also been shown to produce anxiolytic effects during the acute phase of treatment.18, 101, 102, 103 Indeed, administration of either of these two drugs produced an anxiolytic-like effect in HABs together with increased neurogenesis while failing to increase neurogenesis in NABs. To substantiate this finding, application of a selective anxiolytic drug such as a benzodiazepine would be interesting; however, it was not possible to distinguish their anxiolytic from the sedative effect in HAB mice (R Landgraf, personal communication; see comment21). Nevertheless, these findings suggest that the anxiolytic/AD drugs such as L-822,429 and tianeptine drugs may reverse a reduced level of neurogenesis, but do not increase it beyond basal levels as recently suggested.46
Similar findings have also been observed in different animal models of state anxiety/depression for instance, in a corticosterone-induced animal model of anxiety/depression in which fluoxetine did not produce changes in neurogenesis or anxiety in control CD-1 mice, but only in corticosterone-infused mice.40 Also, in another study, stressed tree shrews showed reduced proliferation, which was normalized by tianeptine, while no effect was seen in unstressed tree shrews.58
Our data suggesting that neurogenesis is linked to higher anxiety rather than to depression is supported by results obtained in transgenic mouse line.104 Also studies involving chronic corticosterone administration40 or unpredictable chronic mild stress39 in rodents, leading to the development of higher state anxiety- and depression-like behavior, have suggested that neurogenesis may be associated more with the anxiety-related behavior. These studies together with this study show that neurogenesis appears to be associated with pathological anxiety in animal models of both higher trait and state anxiety and with comorbid depression-related behavior. Taken together, these studies stress the importance of the role of neurogenesis in psychopathological, rather than basal physiological, conditions,36 furthering the ‘neurogenic reserve’ hypothesis, namely, that adult newborn hippocampal neurons,105 which are not essential under normal conditions, comprise a reserve that becomes vital when the organism is faced with stressors.105 HABs with a lower rate of neurogenesis and reduced functional integration of newborn hippocampal neurons would thus be at risk having a lower reserve. In the present context, alterations in the new hippocampal neurons might thus be particularly critical for anxiety-related behavior.
We further address the fact that, during the first few days of treatment with fluoxetine, but not with L-822,429 or tianeptine, we observed a weight decline. This is consistent with a recent study demonstrating a similar effect in humans that is limited to the acute phase of fluoxetine treatment.106 However, it is unlikely that reduced neurogenesis that was observed was due to weight loss, since body weight reduction as a result of dietary restriction has been shown to increase hippocampal neurogenesis in mice and rats,107, 108, 109 whereas a high-fat diet (leading to an increase in body weight) reduces this effect.110, 111
Hypothalamo-pituitary axis dysregulation has been observed both in depressed patients15 as well as animal models of depression32, 36 and ADs have been shown to modulate such alterations,36 detailed investigation of the hypothalamo-pituitary axis would be a desirable next step in this line of research.
Conclusion
This study makes several contributions to our current understanding of the neuropathology underlying comorbid anxiety and higher depression-like behavior. First, it is shown that hippocampal neurogenesis is reduced and functional integration of newly born neurons is impaired in HAB mice with a genetic predisposition to hyper-anxiety and depression-like behavior. Second, hyper-anxiety is specifically associated with hippocampal neurogenesis, whereas modulation of higher depression-like behavior seems to be regulated by neurogenesis-independent mechanisms. Third, after fluoxetine discontinuation remission from higher depression-like behavior can be achieved for several weeks before the onset of relapse. Reversal of emotional challenge-induced DG hypoactivity parallels all the behavioral changes and is thus suggested as a neurobiological marker for successful behavioral remission. Finally, the present data raise the possibility that low-dose maintenance therapy might be necessary for achieving complete remission of higher depression-like behavior in this animal model.
References
Klenk MM, Strauman TJ, Higgins ET . Regulatory focus and anxiety: a self-regulatory model of GAD-depression comorbidity. Pers Indiv Differ 2011; 50: 935–943.
Fava M, Davidson KG . Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19: 179–200.
Fava M . Diagnosis and definition of treatment-resistant depression. Biol Psychiatry 2003; 53: 649–659.
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163: 28–40.
Barr LC, Heninger GR, Goodman W, Charney DS, Price LH . Effects of fluoxetine administration on mood response to tryptophan depletion in healthy subjects. Biol Psychiatry 1997; 41: 949–954.
Gelfin Y, Gorfine M, Lerer B . Effect of clinical doses of fluoxetine on psychological variables in healthy volunteers. Am J Psychiatry 1998; 155: 290–292.
Yeragani VK, Pohl R, Mallavarapu M, Balon R . Approximate entropy of symptoms of mood: an effective technique to quantify regularity of mood. Bipolar Disord 2003; 5: 279–286.
Berton O, Nestler EJ . New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci 2006; 7: 137–151.
Landgraf R, Kessler MS, Bunck M, Murgatroyd C, Spengler D, Zimbelmann M et al. Candidate genes of anxiety-related behavior in HAB/LAB rats and mice: focus on vasopressin and glyoxalase-I. Neurosci Biobehav Rev 2007; 31: 89–102.
Sartori SB, Landgraf R, Singewald N . The clinical implications of mouse models of enhanced anxiety. Future neurology 2011; 6: 531–571.
Singewald N . Altered brain activity processing in high-anxiety rodents revealed by challenge paradigms and functional mapping. Neurosci Biobehav Rev 2007; 31: 18–40.
Muigg P, Scheiber S, Salchner P, Bunck M, Landgraf R, Singewald N . Differential stress-induced neuronal activation patterns in mouse lines selectively bred for high, normal or low anxiety. PloS one 2009; 4: e5346.
Fava GA . Can long-term treatment with antidepressant drugs worsen the course of depression? J Clin Psychiatry 2003; 64: 123–133.
Altshuler L, Suppes T, Black D, Nolen WA, Keck PE, Frye MA et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry 2003; 160: 1252–1262.
Palazidou E . The neurobiology of depression. Br Med Bull 2012; 101: 127–145.
Geddes JR, Carney SM, Davies C, Furukawa TA, Kupfer DJ, Frank E et al. Relapse prevention with antidepressant drug treatment in depressive disorders: a systematic review. Lancet 2003; 361: 653–661.
Samuels BA, Leonardo ED, Gadient R, Williams A, Zhou J, David DJ et al. Modeling treatment-resistant depression. Neuropharmacology 2011; 61: 408–413.
McEwen BS, Chattarji S, Diamond DM, Jay TM, Reagan LP, Svenningsson P et al. The neurobiological properties of tianeptine (Stablon): from monoamine hypothesis to glutamatergic modulation. Mol Psychiatry 2010; 15: 237–249.
Jay T . Cellular plasticity and the pathophysiology of depression. In: Neuroplasticity (Costa e Silva JA et al. (eds). Springer Healthcare Ltd, 2011, pp 41–55.
Kasper S, Olie JP . A meta-analysis of randomized controlled trials of tianeptine versus SSRI in the short-term treatment of depression. Eur Psychiatry 2002; 17 (Suppl 3): 331–340.
Sartori SB, Hauschild M, Bunck M, Gaburro S, Landgraf R, Singewald N . Enhanced fear expression in a psychopathological mouse model of trait anxiety: pharmacological interventions. PLoS One 2011; 6: e16849.
Ebner K, Sartori SB, Singewald N . Tachykinin receptors as therapeutic targets in stress-related disorders. Curr Pharm Des 2009; 15: 1647–1674.
Ratti E, Bellew K, Bettica P, Bryson H, Zamuner S, Archer G et al. Results from 2 randomized, double-blind, placebo-controlled studies of the novel NK1 receptor antagonist casopitant in patients with major depressive disorder. J Clin Psychopharmacol 2011; 31: 727–733.
Lupien SJ, McEwen BS, Gunnar MR, Heim C . Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci 2009; 10: 434–445.
Amaral DG, Witter MP . The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience 1989; 31: 571–591.
Floresco SB, Todd CL, Grace AA . Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci 2001; 21: 4915–4922.
Kingwell K . Depression: In pursuit of happiness. Nat Rev Neurosci 2010; 11: 786.
Kempton MJ, Salvador Z, Munafo MR, Geddes JR, Simmons A, Frangou S et al. Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Arch Gen Psychiatry 2011; 68: 675–690.
Bjornebekk A, Mathe AA, Brene S . The antidepressant effect of running is associated with increased hippocampal cell proliferation. Int J Neuropsychopharmacol 2005; 8: 357–368.
Bjornebekk A, Mathe AA, Brene S . Running has differential effects on NPY, opiates, and cell proliferation in an animal model of depression and controls. Neuropsychopharmacology 2006; 31: 256–264.
Lucassen PJ, Bosch OJ, Jousma E, Kromer SA, Andrew R, Seckl JR et al. Prenatal stress reduces postnatal neurogenesis in rats selectively bred for high, but not low, anxiety: possible key role of placental 11beta-hydroxysteroid dehydrogenase type 2. Eur J Neurosci 2009; 29: 97–103.
Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA . Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011; 476: 458–461.
Airan RD, Meltzer LA, Roy M, Gong Y, Chen H, Deisseroth K . High-speed imaging reveals neurophysiological links to behavior in an animal model of depression. Science 2007; 317: 819–823.
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003; 301: 805–809.
Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G et al. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry 2008; 64: 293–301.
Surget A, Tanti A, Leonardo ED, Laugeray A, Rainer Q, Touma C et al. Antidepressants recruit new neurons to improve stress response regulation. Mol Psychiatry 2011; 16: 1177–1188.
Tfilin M, Sudai E, Merenlender A, Gispan I, Yadid G, Turgeman G . Mesenchymal stem cells increase hippocampal neurogenesis and counteract depressive-like behavior. Mol Psychiatry 2010; 15: 1164–1175.
Perera TD, Dwork AJ, Keegan KA, Thirumangalakudi L, Lipira CM, Joyce N et al. Necessity of hippocampal neurogenesis for the therapeutic action of antidepressants in adult nonhuman primates. PloS one 2011; 6: e17600.
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA et al. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry 2009; 14: 764–773, 739.
David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 2009; 62: 479–493.
Holick KA, Lee DC, Hen R, Dulawa SC . Behavioral effects of chronic fluoxetine in BALB/cJ mice do not require adult hippocampal neurogenesis or the serotonin 1 A receptor. Neuropsychopharmacology 2008; 33: 406–417.
Huang GJ, Bannerman D, Flint J . Chronic fluoxetine treatment alters behavior, but not adult hippocampal neurogenesis, in BALB/cJ mice. Mol Psychiatry 2008; 13: 119–121.
Sahay A, Scobie KN, Hill AS, O’Carroll CM, Kheirbek MA, Burghardt NS et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 2011; 472: 466–470.
Singer BH, Jutkiewicz EM, Fuller CL, Lichtenwalner RJ, Zhang H, Velander AJ et al. Conditional ablation and recovery of forebrain neurogenesis in the mouse. J Comp Neurol 2009; 514: 567–582.
Vollmayr B, Simonis C, Weber S, Gass P, Henn F . Reduced cell proliferation in the dentate gyrus is not correlated with the development of learned helplessness. Biol Psychiatry 2003; 54: 1035–1040.
Petrik D, Lagace DC, Eisch AJ . The neurogenesis hypothesis of affective and anxiety disorders: are we mistaking the scaffolding for the building? Neuropharmacology 2012; 62: 21–34.
Sahay A, Hen R . Adult hippocampal neurogenesis in depression. Nat Neurosci 2007; 10: 1110–1115.
Tanti A, Belzung C . Open questions in current models of antidepressant action. Br J Pharmacol 2010; 159: 1187–1200.
Jessberger S, Kempermann G . Adult-born hippocampal neurons mature into activity-dependent responsiveness. Eur J Neurosci 2003; 18: 2707–2712.
Mayberg HS, Brannan SK, Tekell JL, Silva JA, Mahurin RK, McGinnis S et al. Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry 2000; 48: 830–843.
Stone EA, Lin Y, Quartermain D . A final common pathway for depression? Progress toward a general conceptual framework. Neurosci Biobehav Rev 2008; 32: 508–524.
Marcus SM, Young EA, Kerber KB, Kornstein S, Farabaugh AH, Mitchell J et al. Gender differences in depression: findings from the STAR*D study. J Affect Disord 2005; 87: 141–150.
Dalla C, Pitychoutis PM, Kokras N, Papadopoulou-Daifoti Z . Sex differences in animal models of depression and antidepressant response. Basic Clin Pharmacol Toxicol 2010; 106: 226–233.
Workman JL, Weber MD, Nelson RJ . Dietary arginine depletion reduces depressive-like responses in male, but not female, mice. Behav Brain Res 2011; 223: 81–87.
Kee N, Teixeira CM, Wang AH, Frankland PW . Imaging activation of adult-generated granule cells in spatial memory. Nat Protoc 2007; 2: 3033–3044.
Lehner B, Sandner B, Marschallinger J, Lehner C, Furtner T, Couillard-Despres S et al. The dark side of BrdU in neural stem cell biology: detrimental effects on cell cycle, differentiation and survival. Cell Tissue Res 2011; 345: 313–328.
Singewald N, Chicchi GG, Thurner CC, Tsao KL, Spetea M, Schmidhammer H et al. Modulation of basal and stress-induced amygdaloid substance P release by the potent and selective NK1 receptor antagonist L-822429. J Neurochem 2008; 106: 2476–2488.
Czeh B, Michaelis T, Watanabe T, Frahm J, de Biurrun G, van Kampen M et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci USA 2001; 98: 12796–12801.
Sartori SB, Whittle N, Hetzenauer A, Singewald N . Magnesium deficiency induces anxiety and HPA axis dysregulation: modulation by therapeutic drug treatment. Neuropharmacology 2012; 62: 304–312.
Busquet P, Nguyen NK, Schmid E, Tanimoto N, Seeliger MW, Ben-Yosef T et al. CaV1.3 L-type Ca2+ channels modulate depression-like behaviour in mice independent of deaf phenotype. Int J Neuropsychopharmacol 2010; 13: 499–513.
Strekalova T, Spanagel R, Bartsch D, Henn FA, Gass P . Stress-induced anhedonia in mice is associated with deficits in forced swimming and exploration. Neuropsychopharmacology 2004; 29: 2007–2017.
Couillard-Despres S, Wuertinger C, Kandasamy M, Caioni M, Stadler K, Aigner R et al. Ageing abolishes the effects of fluoxetine on neurogenesis. Mol Psychiatry 2009; 14: 856–864.
Klempin F, Babu H, De Pietri Tonelli D, Alarcon E, Fabel K, Kempermann G . Oppositional effects of serotonin receptors 5-HT1a, 2, and 2c in the regulation of adult hippocampal neurogenesis. Front Mol Neurosci 2010; 3.
Mayhew TM, Mwamengele GL, Dantzer V . Stereological and allometric studies on mammalian cerebral cortex with implications for medical brain imaging. J Anat 1996; 189 (Pt 1): 177–184.
Stemberger S, Poewe W, Wenning GK, Stefanova N . Targeted overexpression of human alpha-synuclein in oligodendroglia induces lesions linked to MSA-like progressive autonomic failure. Exp Neurol 2010; 224: 459–464.
Nutt DJ . Overview of diagnosis and drug treatments of anxiety disorders. CNS Spectr 2005; 10: 49–56.
Salchner P, Singewald N . Neuroanatomical substrates involved in the anxiogenic-like effect of acute fluoxetine treatment. Neuropharmacology 2002; 43: 1238–1248.
Silva MT, Alves CR, Santarem EM . Anxiogenic-like effect of acute and chronic fluoxetine on rats tested on the elevated plus-maze. Brazilian J Med Biol Res=Revista brasileira de pesquisas medicas e biologicas/ Sociedade Brasileira de Biofisica [et al] 1999; 32: 333–339.
Gaburro S, Stiedl O, Giusti P, Sartori SB, Landgraf R, Singewald N . A mouse model of high trait anxiety shows reduced heart rate variability that can be reversed by anxiolytic drug treatment. Int J Neuropsychopharmacol 2011; 14: 1341–1355.
Treadway MT, Zald DH . Reconsidering anhedonia in depression: lessons from translational neuroscience. Neurosci Biobehav Rev 2011; 35: 537–555.
Bunck M, Czibere L, Horvath C, Graf C, Frank E, Kessler MS et al. A hypomorphic vasopressin allele prevents anxiety-related behavior. PloS one 2009; 4: e5129.
Kromer SA, Kessler MS, Milfay D, Birg IN, Bunck M, Czibere L et al. Identification of glyoxalase-I as a protein marker in a mouse model of extremes in trait anxiety. J Neurosci 2005; 25: 4375–4384.
Sibille E, Wang Y, Joeyen-Waldorf J, Gaiteri C, Surget A, Oh S et al. A molecular signature of depression in the amygdala. Am J Psychiatry 2009; 166: 1011–1024.
Chambers CD, Johnson KA, Dick LM, Felix RJ, Jones KL . Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996; 335: 1010–1015.
Benninghoff J, van der Ven A, Schloesser RJ, Moessner R, Moller HJ, Rujescu D . The complex role of the serotonin transporter in adult neurogenesis and neuroplasticity. A critical review. World J Biol Psychiatry 2012; 13: 240–247.
Zhao Z, Baros AM, Zhang HT, Lapiz MD, Bondi CO, Morilak DA et al. Norepinephrine transporter regulation mediates the long-term behavioral effects of the antidepressant desipramine. Neuropsychopharmacology 2008; 33: 3190–3200.
Zhao Z, Zhang HT, Bootzin E, Millan MJ, O’Donnell JM . Association of changes in norepinephrine and serotonin transporter expression with the long-term behavioral effects of antidepressant drugs. Neuropsychopharmacology 2009; 34: 1467–1481.
Oh JE, Zupan B, Gross S, Toth M . Paradoxical anxiogenic response of juvenile mice to fluoxetine. Neuropsychopharmacology 2009; 34: 2197–2207.
Dagyte G, Trentani A, Postema F, Luiten PG, Den Boer JA, Gabriel C et al. The novel antidepressant agomelatine normalizes hippocampal neuronal activity and promotes neurogenesis in chronically stressed rats. CNS Neurosci Ther 2010; 16: 195–207.
Huang YH, Cheng CY, Hong CJ, Tsai SJ . Expression of c-Fos-like immunoreactivity in the brain of mice with learned helplessness. Neurosci Lett 2004; 363: 280–283.
Lee BT, Seong Whi C, Hyung Soo K, Lee BC, Choi IG, Lyoo IK et al. The neural substrates of affective processing toward positive and negative affective pictures in patients with major depressive disorder. Prog Neuro-Psych Biol Psych 2007; 31: 1487–1492.
Milne AM, Macqueen GM, Hall GB . Abnormal hippocampal activation in patients with extensive history of major depression: an fMRI study. J Psychiatry Neurosci 2012; 37: 28–36.
Kuhn S, Gallinat J . Resting-state brain activity in schizophrenia and major depression: a quantitative meta-analysis. Schizophrenia Bulletin, advance online publication, 10 November 2011 (doi:10.1093/schbul/sbr151).
Drew MR, Hen R . Adult hippocampal neurogenesis as target for the treatment of depression. CNS Neurol Disord Drug Targets 2007; 6: 205–218.
Sapolsky RM . Is impaired neurogenesis relevant to the affective symptoms of depression? Biol Psychiatry 2004; 56: 137–139.
Zhao C, Deng W, Gage FH . Mechanisms and functional implications of adult neurogenesis. Cell 2008; 132: 645–660.
Taliaz D, Stall N, Dar DE, Zangen A . Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry 2010; 15: 80–92.
Pittenger C, RS Duman . Stress depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 2008; 33: 88–109.
Kee N, Teixeira CM, Wang AH, Frankland PW . Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat Neurosci 2007; 10: 355–362.
Snyder JS, Choe JS,, Clifford MA, Jeurling SI, Hurley P, Brown A et al. Adult-born hippocampal neurons are more numerous, faster maturing, and more involved in behavior in rats than in mice. J Neurosci 2009; 29: 14484–14495.
Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A et al. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry 2006; 11: 514–522.
Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J et al. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009; 34: 2376–2389.
Perera TD, Coplan JD, Lisanby SH, Lipira CM, Arif M, Carpio C et al. Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J Neurosci 2007; 27: 4894–4901.
DeCarolis NA, Eisch AJ . Hippocampal neurogenesis as a target for the treatment of mental illness: a critical evaluation. Neuropharmacology 2010; 58: 884–893.
Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG . Transient expression of doublecortin during adult neurogenesis. J Comp Neurol 2003; 467: 1–10.
Andrews PW, Thomson JA, Amstadter A, Neale MC . Primum non nocere: an evolutionary analysis of whether antidepressants do more harm than good. Front Psychol 2012; 3: 117.
Kalia M, O’Callaghan JP, Miller DB, Kramer M . Comparative study of fluoxetine, sibutramine, sertraline and dexfenfluramine on the morphology of serotonergic nerve terminals using serotonin immunohistochemistry. Brain Res 2000; 858: 92–105.
Stergiou L, Hengartner MO . Death and more: DNA damage response pathways in the nematode C. elegans. Cell Death Differ 2004; 11: 21–28.
Wu X, Castren E . Co-treatment with diazepam prevents the effects of fluoxetine on the proliferation and survival of hippocampal dentate granule cells. Biol Psychiatry 2009; 66: 5–8.
Kempermann G, Kuhn HG, Gage FH . Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc Nat Acad Sci Usa 1997; 94: 10409–10414.
Ebner K, Singewald GM, Whittle N, Ferraguti F, Singewald N . Neurokinin 1 receptor antagonism promotes active stress coping via enhanced septal 5-HT transmission. Neuropsychopharmacology 2008; 33: 1929–1941.
File SE, Andrews N, al-Farhan M . Anxiogenic responses of rats on withdrawal from chronic ethanol treatment: effects of tianeptine. Alcohol Alcohol 1993; 28: 281–286.
Zethof TJ, Van der Heyden JA, Tolboom JT, Olivier B . Stress-induced hyperthermia as a putative anxiety model. Eur J Pharmacol 1995; 294: 125–135.
Revest JM, Dupret D, Koehl M, Funk-Reiter C, Grosjean N, Piazza PV et al. Adult hippocampal neurogenesis is involved in anxiety-related behaviors. Mol Psychiatry 2009; 14: 959–967.
Kempermann G . The neurogenic reserve hypothesis: what is adult hippocampal neurogenesis good for? Trends Neurosci 2008; 31: 163–169.
Serretti A, Mandelli L . Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry 2010; 71: 1259–1272.
Lee J, Duan W, Long JM, Ingram DK, Mattson MP . Dietary restriction increases the number of newly generated neural cells, and induces BDNF expression, in the dentate gyrus of rats. J Mol Neurosci 2000; 15: 99–108.
Lee J, Duan W, Mattson MP . Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem 2002; 82: 1367–1375.
Lee J, Seroogy KB, Mattson MP . Dietary restriction enhances neurotrophin expression and neurogenesis in the hippocampus of adult mice. J Neurochem 2002; 80: 539–547.
Park HR, Lee J . Neurogenic contributions made by dietary regulation to hippocampal neurogenesis. Ann N Y Acad Sci 2011; 1229: 23–28.
Park HR, Park M, Choi J, Park KY, Chung HY, Lee J . A high-fat diet impairs neurogenesis: involvement of lipid peroxidation and brain-derived neurotrophic factor. Neurosci Lett 2010; 482: 235–239.
Acknowledgements
This work was supported by research grants from the Austrian Science Fund (FWF), W 1206-B18 SPIN (to NS and LK) and SFB F44 (to NS and LA) and Hope for Depression Research/ISAN (to NS). We thank Markus Hauschild for excellent technical assistance, Christina Bachmann for excellent animal care and Servier Austria GmbH for their kind gift of tianeptine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Sah, A., Schmuckermair, C., Sartori, S. et al. Anxiety- rather than depression-like behavior is associated with adult neurogenesis in a female mouse model of higher trait anxiety- and comorbid depression-like behavior. Transl Psychiatry 2, e171 (2012). https://doi.org/10.1038/tp.2012.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2012.94
Keywords
This article is cited by
-
Neuroinflammatory alterations in trait anxiety: modulatory effects of minocycline
Translational Psychiatry (2020)
-
Modelling depression in animals: at the interface of reward and stress pathways
Psychopharmacology (2017)
-
Selective Mitochondrial Targeting Exerts Anxiolytic Effects In Vivo
Neuropsychopharmacology (2016)
-
Treatment-resistant depression: are animal models of depression fit for purpose?
Psychopharmacology (2015)
-
Anhedonic behavior in cryptochrome 2-deficient mice is paralleled by altered diurnal patterns of amygdala gene expression
Amino Acids (2015)
