
Abstract
Nickel-based hydroxide hierarchical nanoarrays (NiyM(OH)x HNAs M = Fe or Zn) are doped with non-noble transition metals to create nanostructures and regulate their activities for the oxygen evolution reaction. Catalytic performance in these materials depends on their chemical composition and the presence of nanostructures. These novel hierarchical nanostructures contain small secondary nanosheets that are grown on the primary nanowire arrays, providing a higher surface area and more efficient mass transport for electrochemical reactions. The activities of the NiyM(OH)x HNAs for the oxygen evolution reaction (OER) followed the order of Ni2.2Fe(OH)x > Ni(OH)2 > Ni2.1Zn(OH)x, and these trends are supported by density functional theory (DFT) calculations. The Fe-doped nickel hydroxide hierarchical nanoarrays (Ni2.2Fe(OH)x HNAs), which had an appropriate elemental composition and hierarchical nanostructures, achieve the lowest onset overpotential of 234 mV and the smallest Tafel slope of 64.3 mV dec−1. The specific activity, which is normalized to the Brunauer–Emmett–Teller (BET) surface area of the catalyst, of the Ni2.2Fe(OH)x HNAs is 1.15 mA cm−2BET at an overpotential of 350 mV. This is ~4-times higher than that of Ni(OH)2. These values are also superior to those of a commercial IrOx electrocatalyst.
Similar content being viewed by others


Introduction
Because of growing demands for energy and environmental concerns, advanced technologies are being sought for the production of inexpensive, sustainable, and carbon-neutral fuels1,2,3,4,5. Water splitting is promising for the production of fuel from renewable but intermittent energy sources, such as wind and solar6. The anodic water-splitting reaction, the oxygen evolution reaction (OER), is a significant source of efficiency losses, because it involves multistep proton-coupled electron transfer and sluggish kinetics7. Currently, noble metal-based compounds, such as IrOx and RuO2, are the most active OER catalysts, but their scarcity and high cost limit their use in practical applications8,9,10,11. Therefore, alternative electrode configurations are needed with extraordinary activities and superior long-term stabilities for the OER12,13,14.
Low-cost transition metal catalysts, especially Ni15,16, have been studied extensively, exhibit good catalytic activities, and are stable against corrosion during the OER in alkaline media. The development of improved Ni-based OER catalysts can be accelerated by an improved understanding of the intrinsic catalytic activity of materials and their dependence on catalyst composition and structure. The OER activity of Ni-based catalysts can be significantly enhanced by doping with other earth-abundant elements such as Co, Fe, Mn, and Zn17,18,19,20,21,22. Therefore, a systematic study of the OER activities achieved when different dopants are added to Ni-based hydroxides would provide valuable insight into the synthesis of improved OER catalysts23. Recently, experimental studies suggest that the OER activities of oxyhydroxide thin films follow the order of Ni(Fe)OxHy > Co(Fe)OxHy > FeOxHy-AuOx > FeOxHy > CoOxHy > NiOxHy > MnOxHy24. Experimental studies have shown that the overpotentials required to achieve 10 mA cm−2 for heterogeneous electrocatalysts follow the order of NiFeOx < CoFeOx < NiCoOx CoOx < NiLaOx < NiCuOx < CoOx/CoPi < NiOx < NiCeOx25. However, the performance of OER catalysts can be limited by the low conductivities24 and surface areas25 of ordinary nanostructures, particularly those containing nanoparticles. Previous studies have shown that the OER activity of an ultra-thin γ-CoOOH nanosheet is 20-times higher than that of bulk CoOOH and 2.4-times higher than that of an IrO2 electrocatalyst26. Well-aligned nanowire arrays have been used as highly effective electrodes, because of their intrinsic advantages27,28,29. Moreover, ordinary electrodes have relatively poor stabilities because of contact between the substrate and electrocatalysts during the electrocatalytic of OER, especially at large current densities. Therefore, hierarchically architectures can be constructed on conductive metal substrates to form high-performance nanocatalysts electrode30,31. However, no systematic studies have been performed that combine theoretical and experimental characterizations of the relationship between the doping of transition metals and the OER activities of Ni-based hydroxide nanoarrays.
In this work, the nanostructured morphology and OER activity of Ni-based hydroxide hierarchical nanoarrays (NiyM(OH)x HNAs, M = Fe or Zn) were modified using two non-noble transition metals (Fe and Zn) as dopants. A systematic experimental and theoretical study of the effect of transition-metal doping on the nanostructure and OER activity of nickel-based catalysts is presented in this work. The intrinsic OER activity trends of the NiyM(OH)x HNAs followed Ni2.2Fe(OH)x > Ni(OH)2 > Ni2.1Zn(OH)x. Theoretical and experimental results were in good agreement. The trends were explained in terms of the surface areas and compositions of active sites, providing potential insights for the future design of more efficient water-splitting catalysts.
Results and Discussion
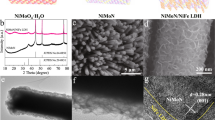
NiyM(OH)x HNAs, where M = Fe and Zn, were fabricated by dipping Cu foam substrates coated with one-dimensional (1D) Cu2O nanowire arrays into an aqueous solution containing metal chloride salts and sodium hyposulfite using a solution-phase cation exchange method at room temperature. During the cation exchange process, the Cu2O nanowires were etched by S2O32−, releasing OH−. During this process NiyM(OH)x HNAs precipitated, these new NiyM(OH)x HNAs structures inherited the geometry of the Cu2O template. Secondary NiyM(OH)x HNAs nanostructures also formed depending on the solubility of the products and the pH of the reaction system. As illustrated in Fig. 1, the secondary nanostructures of the NiyM(OH)x HNAs were regulated during this process. Low magnification SEM images of the NiyM(OH)x HNAs revealed that the surface of the Cu foam substrate was completely covered with vertically aligned nanoarrays (Fig. 2). The inset to Fig. 2a showed the morphology of the Ni(OH)2 HNAs, which inherited the shape of the 1D Cu2O nanowire arrays (Fig. S1) along the axial direction. After doping Ni(OH)2 with transition metals, the surfaces of the nanowires became rougher, and their morphologies markedly changed into hierarchical structures with secondary nanosheets grown on the primary nanowire arrays (see insets to Fig. 2b,c). The degree of surface roughness on the NiyM(OH)x HNAs followed the order of Ni(OH)2 < Ni2.1Zn(OH)x < Ni2.2Fe(OH)x, indicating a marked increasement in surface area when the appropriate elements were used as dopants. Transmission electron microscopy (TEM) images of the NiyM(OH)x HNAs further revealed the presence of secondary nanosheets (Figs 3 and S2). The Ni2.2Fe(OH)x HNAs had the most irregularly shaped nanosheet coating. As shown in Fig. S3, the Ni(OH)2, Ni2.1Zn(OH)x, and Ni2.2Fe(OH)x HNAs had diameters of 30.5 nm, 51 nm, and 103.4 nm, respectively. Cross-sectional SEM images of the NiyM(OH)x HNAs (Fig. S4) revealed that the three NiyM(OH)x HNAs had similar lengths of 2 μm. Figure 3c,d show high-magnification and high-resolution TEM (HRTEM) images of the Ni2.2Fe(OH)x HNAs. Ultra-thin (~2.1 nm) character was clearly observed on one edge curled nanosheet of the Ni2.2Fe(OH)x HNAs, indicating more exposure of low coordinated surface atoms and thus abundant catalytically active sites. HRTEM images indicated that the Ni(OH)2 HNAs were predominantly crystalline, while the Ni2.2Fe(OH)x and Ni2.1Zn(OH)x HNAs were amorphous (Figs S2b, 3d and S2e). Fast Fourier transform (FFT) images were in agreement with the HRTEM images. SEM and TEM results indicated that the proposed method effectively regulated the growth of nanostructures on the Ni(OH)2 HNAs using Fe and Zn as dopants. When doping with the transition metals Fe and Zn, the surfaces of the HNAs become rougher and more highly amorphous. Figure S5 shows X-ray diffraction (XRD) patterns for the Cu2O nanowire arrays and the CoyFe1-y(OH)x HNAs. The diffraction patterns for the Cu2O nanowire arrays indicated the presence of Cu2O phases (PDF#65-3288) and Cu (PDF# 65-9743). The diffraction patterns for the Ni(OH)2, Ni2.1Zn(OH)x, and Ni2.2Fe(OH)x HNAs did not contain any characteristic peaks for Ni, Fe, or Zn compounds. Only Cu and a small amount of Cu2O were present (Fig. S5), revealing that the three samples had amorphous structures. It should be noted that the amorphous nature of the Ni(OH)2 HNAs observed by XRD did not conflict with the crystal structures obtained from HRTEM, because the faint crystal lattice and weak FFT pattern of the Ni(OH)2 HNAs indicated a low crystallinity32,33.

Depiction of the growth of Zn- and Fe-doped NiyM(OH)x HNAs from sacrificial Cu2O nanoarray templates.

Low- and high-magnification (inset) SEM images of the (a) Ni(OH)2, (b) Ni2.2Fe(OH)x, and (c) Ni2.1Zn(OH)x HNAs.

(a,b) TEM images, (c) high-magnification TEM image, and (d) HRTEM and FFT (inset) images of the Ni2.2Fe(OH)x HNAs.
The Ni/M atomic ratios of the NiyM(OH)x HNAs were determined with inductively coupled plasma (ICP) emission spectrometry. The ratios in the HNAs were similar to the reactant ratios (Table S1), indicating that the Ni/M ratios in the hydroxides were similar to those in the precursors. The surface compositions and valence states of the as-prepared NiyM(OH)x HNAs were investigated by X-ray photoelectron spectroscopy (XPS), and the results are shown in Fig. 4. Nickel, Fe, Zn, and O species were observed. Peak fitting analysis of the Ni 2p for Ni(OH)2 revealed one Ni2+ state for Ni at binding energies of 855.5 eV and 873.5 eV. Peak fitting analysis for Ni 2p in the Ni2.2Fe(OH)x and Ni2.1Zn(OH)x HNAs indicated the presence of Ni2+ (855.4 eV and 873.1 eV) and Ni3+ (857.3 eV and 875.5 eV)34. Compared to the un-doped Ni(OH)2 HNAs, the Ni 2p peaks of the M-doped samples were shifted to more positive energies (Ni2.1Zn(OH)x < Ni2.2Fe(OH)x), suggesting that the oxidation of Ni2+ was favored when Fe and Zn were added. This effect was strongest with Fe35. Additional evidence for the presence of Ni2+ was observed in the two intense shakeup satellite peaks (861.8 eV and 880.0 eV)16. The Zn 2p XPS spectrum for the Ni2.1Zn(OH)x HNAs contained 2p3/2 and 2p1/2 doublets, which are characteristic of Zn2+ (1022.7 eV and 1045.7 eV)34. Fe 2p3/2 and Fe 2p1/2 spin-orbital splitting for the Ni2.2Fe(OH)x HNAs was deconvolved into four peaks, indicating the coexistence of Fe2+ (711.5 eV and 723.7 eV) and Fe3+ (716.0 eV and 726.3 eV) in the Ni2.2Fe(OH)x HNAs36,37. The O 1s spectrum of the Ni(OH)2 HNAs was fit to a peak at a binding energy of 530.9 eV, which was assigned to the oxygen in hydroxide. The O 1s spectra of the Ni2.2Fe(OH)x and Ni2.1Zn(OH)x HNAs were fit with two peaks at binding energies of 530.1 eV and 531 eV, revealing the presence of lattice and hydroxide oxygens, respectively16. These results confirm that strong electron interactions occurred between Ni and both Fe and Zn in the NiyM(OH)x HNAs.

High-resolution XPS spectra of the (a) Ni 2p and (b) O 1s regions for the Ni(OH)2, Ni2.1Zn(OH)x, and Ni2.2Fe(OH)x HNAs (from bottom to top); (c) Zn 2p spectra of the Ni2.1Zn(OH)x HNAs, (d) Fe 2p spectra of the Ni2.2Fe(OH)x HNAs.
The effect of doping the NiyM(OH)x HNAs on their electrochemical behaviors were investigated using cyclic voltammetry (CV) in 1 M KOH. As shown in Fig. 5a, the CV curves of all three electrodes exhibited coupled redox peaks. The redox couple (1.45 V/1.27 V vs. RHE) of the Ni(OH)2 HNAs corresponded to the transformation between Ni(OH)2 and NiOOH16. The Ni2.1Zn(OH)x and Ni2.2Fe(OH)x HNAs’ exhibited redox peaks at 1.43 V/1.34 V vs. RHE and 1.44 V/1.35 V vs. RHE, respectively. These peaks corresponded to the transformation between Ni(OH)2 and NiOOH, and the positive shifts in redox potential were caused by adding the dopants (Fe or Zn) to the electrodes19,38.

(a) CV curves of the NiyM(OH)x HNAs. (b) LSV curves of the NiyM(OH)x HNAs, Cu2O nanoarray, and IrOx. (c) BET surface areas (d) LSV curves normalized to the BET surface areas of the NiyM(OH)x HNAs. (e) Tafel plots for the NiyM(OH)x HNAs and IrOx. (f) Chronoamperometric measurements for the NiyM(OH)x HNAs.
The effect of doping the NiyM(OH)x HNAs on their OER catalytic activities were tested with linear sweep voltammetry (LSV) in 1 M KOH (Fig. 5b). The Cu2O nanoarrays exhibited a negligible catalytic activity, while the OER current of the Ni2.2Fe(OH)x HNAs was much higher than those of the other electrodes. The Ni2.2Fe(OH)x HNAs had a low OER onset overpotential (η) of 234 mV, which was more negative than the η of the Ni(OH)2 HNAs (254 mV) and commercial IrOx electrocatalyst (248 mV). The high catalytic activity of the Ni2.2Fe(OH)x HNAs was also indicated by its ability to support a given current density (j) at a lower η than the other electrodes. At j = 100 mA cm−2, the as-prepared Ni2.2Fe(OH)x HNAs required an η of 298 mV, which was 125 mV and 177 mV less than the η values of the Ni(OH)2 HNAs (423 mV) and IrOx (375 mV), respectively. Therefore, intrinsic activities were compared at η = 350 mV. The intrinsic activity of the Ni2.2Fe(OH)x HNAs was 16- and 5-times higher than those of the Ni(OH)2 HNAs and IrOx, respectively, revealing strong interactions between Ni and Fe during OER catalysis. Meanwhile, the Ni2.1Zn(OH)x HNAs exhibits a more positive onset η of 276 mV, and required a high η of 410 mV to achieve a current density of 100 mA cm−2. The intrinsic activity of the Ni2.1Zn(OH)x HNAs (25.8 mA cm−2) was similar to that of the Ni(OH)2 HNAs. These results suggest that unfavorable interactions occurred between Ni and Zn. The excellent OER activities of the catalysts were attributed to their increased surface areas and specific activities (active sites per unit area).
The Brunauer–Emmett–Teller (BET) surface area measurements were performed to confirm the mesoporous nature of the NiyM(OH)x HNAs. Nitrogen adsorption-desorption curves revealed a Type IV isotherm (Fig. S8). Additionally, a H3-type hysteresis loop was observed, providing further evidence of nanosheet aggregation39. As shown in Fig. 5c, the Ni(OH)2 HNAs (73.2 cm2 g−1) had a smaller BET surface area than the Ni2.1Zn(OH)x (105.6 cm2 g−1) and Ni2.2Fe(OH)x (155.6 cm2 g−1) HNAs. These results confirmed observations of increased surface areas in the SEM (Fig. 2) and TEM (Figs 3 and S2) images. Specific activity (current per BET area) is a measure of the density of active sites on the surface of a catalyst. Figure 5d shows LSV curves after normalizing the measured currents to the catalysts’ BET surface areas. The specific activity of the Ni2.2Fe(OH)x HNAs was 1.15 mA cm−2BET at η = 350 mV, which was 4- and 8-times higher than those of the Ni(OH)2 HNAs (0.22 mA cm−2BET) and Ni2.1Zn(OH)x HNAs (0.13 mA cm−2BET), respectively. These results indicated that adding Fe indeed resulted in more active sites, while doping with Zn decreased the number of active sites.
Kinetic analyses were performed using LSV to generate Tafel plots and electrochemical impedance spectra (EIS). As shown in Fig. 5e, the resulting Tafel slope of the Ni2.2Fe(OH)x HNAs was 64.3 mV dec−1, which was much lower than that of the Ni(OH)2 HNAs (123.4 mV dec−1), Ni2.1Zn(OH)x HNAs (107.2 mV dec−1), and IrOx (113.3 mV dec−1). Tafel slopes were used to probe the OER mechanisms of the catalysts. Efficient electron and mass transport result in lower Tafel slopes. The EIS was performed in oxygen-saturated 1.0 M KOH (Fig. S6). ZSimpWin 3.5 (Zolartron Analytical) was used to fit the resistance values, as shown in Table S2. As shown in the inset to Fig. S6, all of the NiyM(OH)x HNAs electrodes were fitted using the same equivalent circuit, which contained three components: solution resistance (Rs), charge-transfer resistance (Rct), and constant-phase resistance (Rcp). The Ni2.2Fe(OH)x HNAs had an Rct of 1.7 Ω, which was much lower than that of the Ni2.1Zn(OH)x (21.4 Ω) and Ni(OH)2 (23.8 Ω) HNAs. These results indicated that OER kinetics were enhanced for the Ni2.2Fe(OH)x HNAs electrode. These EIS measurements were consistent with the findings from LSV.
Turnover frequency (TOF) is an intrinsic property of a catalyst and an important indicator of catalyst performance. The TOF of the Ni2.2Fe(OH)x HNAs was much higher than that of Ni2.1Zn(OH)x and Ni(OH)2 HNAs (Fig. S7). Moreover, at η = 350 mV, the TOF of the Ni2.2Fe(OH)x HNAs was at least 15-, 21-, and 3-times as those of the Ni(OH)2 HNAs (0.011 s–1), the Ni2.1Zn(OH)x HNAs (0.008 s–1), and IrOx (0.05 s–1), respectively. These TOF values further verified the superior catalytic performance of the Ni2.2Fe(OH)x HNAs for the OER. This improved performance resulted from strong interactions between Ni and Fe and the presence of more exposed catalytically active sites.
Long-term stability is also important for catalysts that are to be used for practical applications. As shown in Fig. 5f, all of the NiyM(OH)x HNAs possessed excellent stabilities with nearly no decrease in j after over 20 h of operation in O2-saturated 1 M KOH. This remarkable operational stability was ascribed to the efficiency of the current collector, the material’s excellent intrinsic stability, the robustness of the electrode and a low coverage of gas bubbles on the NiyM(OH)x HNAs40. The higher j values achieved by the Ni2.2Fe(OH)x HNAs was attributed to the presence of hierarchical nanostructures and strong interactions between Ni and Fe. The superior long-term stability of the Ni2.2Fe(OH)x HNAs suggests its potential use as a new electrode in oxygen-evolution devices. The electrochemical properties of NiyM(OH)x and IrOx are summarized in Table 1, and especially the overall performance of Ni2.2Fe(OH)x surpasses most reported typical Co-based electrocatalysts for water oxidation under alkaline solution (Table S3).
To better understand the catalytic activities that resulted from doping with Fe and Zn, the binding energies of oxygen on the NiyM(OH)x catalysts were investigated using DFT calculations. The binding energy of oxygen serves as a reliable measure of the activity of a catalyst toward the OER. Smaller values of binding energy of oxygen(Eo) at a reaction site correspond to higher activities for the OER41,42. Hydroxide clusters were constructed based on a model of Ni2M(OH)6. The oxygen binding energies of the hydroxide clusters increased in the order of Ni2Zn(OH)6 > Ni3(OH)6 > Ni2Fe(OH)6 (Fig. 6b). The reactivities of the amorphous hydroxides followed the order of Ni2Fe(OH)6 > Ni3(OH)6 > Ni2Zn(OH)6, which was in good agreement with the experimental results.

(a) Optimal hydroxide models and adsorption models. Atoms in the clusters: blue = Ni; green = Zn; gray = Fe; red = O; white = H. (b) Eo on the different hydroxide clusters.
Conclusion
Nanostructures were generated to regulate the OER activities of the NiyM(OH)x HNAs using various non-noble transition metals as dopants. According to both experimental and DFT-based theoretical analyses, Fe and Zn had opposite effects on the catalyst’s activity. Fe was an effective dopant, while Zn decreased the OER activity of the catalyst. Hierarchical nanostructures allowed efficient charge transfer and a sufficient surface area for active sites. A 3D porous Cu foam not only provided a large surface area and stable anchoring sites for nanoarrays but also acted as an efficient electron collector. Because of the hierarchical nanostructures, the appropriate elemental composition of the catalyst, and the presence of a multifunctional 3D conductive substrate, the Ni2.2Fe(OH)x HNAs exhibited an enhanced OER activity. The Ni2.2Fe(OH)x HNAs had a low onset η of 234 mV and a small Tafel slope of 64.3 mV dec−1. They also exhibited an excellent long-term stability for over 20 h in an alkaline electrolyte. The Ni2.2Fe(OH)x HNAs also had a superior activity compared to that of a commercial IrOx catalyst, and these Ni2.2Fe(OH)x HNAs were prepared using an extremely simple method. Their activities were comparable to those of other NiFe hydroxides obtained through more labor-intensive procedures. This study provides significant new guidelines for and a broader understanding of the use of dopants to improve catalytic activity.
Methods
Materials synthesis
Nickel(II) dichloride (NiCl2·6H2O, AR), iron (II) dichloride (FeCl2∙4H2O, AR), zinc dichloride (ZnCl2, AR), potassium hydroxide (KOH, AR), sodium hyposulfite (Na2S2O3∙5H2O, AR), oxalic acid (H2C2O4, AR), and ethanol (CH3CH2OH, AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without further purification. Mill-Q water (resistivity > 18 MΩ·cm) was used throughout.
Cu foam (100 pores per inch, 98% porosity, and ~1.5 mm thick) was cut into squares (2.0 cm × 2.0 cm), and cleaned in Mill-Q water and ethanol before use. The Cu foam was then anodized in 0.4 M H2C2O4 for 20 min at 36 V, using a graphite plate cathode. Electro-oxidation was performed with a potentiostat (CHI760D, CH Instruments) in a three-electrode configuration consisting of an anodized Cu foam working electrode, a Pt gauze counter electrode, and an Ag/AgCl reference electrode43. Cyclic voltammetry was performed in the potential range from −0.3 V to 0.1 V at a scan rate of 1 mV s−1 in 1 M KOH for the in situ growth of Cu2O nanowire arrays on the Cu foam43,44.
NiyM(OH)x HNAs were fabricated using the Cu2O nanowire arrays as sacrificial templates33. Briefly, known amounts of NiCl2·6 H2O and MCl2·nH2O (M = Fe or Zn) were dissolved in a mixture containing 17.5 mL Mill-Q water and 17.5 mL ethanol. A 2:1 molar ratio of NiCl2·6H2O and MCl2·nH2O was used to prepare the NiyM(OH)x HNAs. The total moles of NiCl2·6H2O and MCl2·nH2O used was 8 × 10−5. A Cu foam decorated with Cu2O nanowire arrays was immersed in this suspension, which was then stirred at room temperature. Na2S2O3 (1 M, 10 mL) was added dropwise to this mixture under magnetic stirring for 1 h. The substrate was then removed and washed repeatedly in ethanol and Mill-Q water before being dried at 60 °C in a vacuum oven for 4 h.
Structural characterization
Scanning electron microscopy (SEM) was performed with a ZEISS MERLIN scanning electron microscope. Microstructural investigations were performed with a JEOL JEM-2100 and Tecnai G2 Spirit TWIN. X-ray diffraction (XRD) patterns were recorded using a Rigaku Ultima IV. The valence states of elements were measured with X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe). All of the spectra were normalized to the C 1 s binding energy at 284.8 eV. Ni/M atomic ratios were measured with a VISTA-MPX ICP-OES. BET measurements were performed on a Quadrasorb SI analyzer at 77 K.
Electrochemical Measurements
Electrochemical measurements were performed in O2-saturated 1 M KOH with an electrochemical analyzer (CHI760D, CH Instruments), using a three-electrode configuration with an Hg/HgO (1 M KOH) reference electrode that contained a double salt bridge and a platinum mesh counter electrode. The NiyFe1-y(OH)x HNAs (0.5 cm × 0.5 cm) on Cu foams were used as working electrodes. All polarization measurements were performed at a scan rate of 5 mV s−1. Potentials are reported in terms of the reversible hydrogen electrode (RHE), using: E (RHE) = E (Hg/HgO) + 0.098 V + 0.0591 V × pH. All CV measurements were compensated for iR drop by 75%. Stability was measured using the controlled potential electrolysis method. The EIS was performed with a Princeton PMC 1000 electrochemical workstation in the frequency range of 10−2 Hz−104 Hz at an amplitude of 5 mV. All electrochemical tests were performed at 25 °C.
Turnover frequency (TOF) was calculated as: TOF = (j × a)/(4 × n × F), where j is the current density at a given potential, a is the surface area of the electrode (0.25 cm2 for the Cu foam electrode), 4 is the number of electrons transferred in the OER, n is the number of moles of all metal ions available for the OER (including Ni and M), and F is Faraday’s constant (96485 C mol−1).
DFT Calculations
Since the NiyM(OH)x HNAs were predominantly amorphous (the Ni(OH)2 HNAs had a hexagonal Ni(OH)2 phase and the Ni2M(OH)x HNAs had an amorphous phase), cluster rather than slab model was chosen for the DFT simulation. Hydroxide clusters were first built based on the model of Ni2M(OH)645, as shown in Fig. 6a. All reported DFT calculations with the Hubbard U (DFT + U) calculations were performed at the Perdew-Burke-Ernzenhof/Generalized Gradient Approximation (PBE/GGA)46 level using the spin-dependent formulation of the hybrid Gaussian and the plane waves method. The calculations were implemented with the open-source CP2K/QUICKSTEP47,48 code. For a better description of the Ni and Fe 3d electrons, the Hubbard effective terms Ueff(Ni) = 5.96 eV and Ueff(Fe) = 5.3 eV were added to the PBE functional49,50. Electrons in the outer most shells of the atoms were treated as being in their valence states. The Kohn-Sham orbitals of the valence electrons were expanded in molecularly optimized Gaussian basis sets of double-ζ plus polarization quality (MOLOPT-SR-DZVP)51. Ionic cores were represented by norm-conserving Goedecker-Teter-Hutter52,53,54 pseudopotentials. The auxiliary plane wave basis set was truncated with a 500 Ry kinetic energy cut off.
Additional Information
How to cite this article: Zhou, T. et al. Transition metal ions regulated oxygen evolution reaction performance of Ni-based hydroxides hierarchical nanoarrays. Sci. Rep. 7, 46154; doi: 10.1038/srep46154 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Zhang, J., Zhao, Z., Xia, Z. & Dai, L. A metal-free bifunctional electrocatalyst for oxygen reduction and oxygen evolution reactions. Nat. Nanotechnol. 10, 444–452 (2015).
Kim, G., Oh, M. & Park, Y. Solar-rechargeable battery based on photoelectrochemical water oxidation: Solar water battery. Sci. Rep. 6, 33400 (2016).
Wang, W., Zhao, Y. & Ding, Y. 2D ultrathin core-shell Pd@Ptmonolayer nanosheets: defect-mediated thin film growth and enhanced oxygen reduction performance. Nanoscale 7, 11934–11939 (2015).
Lewis, N. S. & Nocera, D. G. Powering the planet: chemical challenges in solar energy utilization. Proc. Nat.l Acad. Sci. USA 103, 15729–15735 (2006).
Kraytsberg, A. & Ein-Eli, Y. Higher, stronger, better a review of 5 volt cathode materials for advanced lithium-ion batteries. Adv. Energy Mater. 2, 922–939 (2012).
Meyer, T. J. The art of splitting water. Nature 451, 778–779 (2008).
Ahn, H. S. & Bard, A. J. Surface Interrogation Scanning Electrochemical Microscopy of Ni(1-x)Fe(x)OOH (0 < x < 0.27) Oxygen Evolving Catalyst: Kinetics of the “fast” Iron Sites. J. Am. Chem. Soc. 138, 313–318 (2016).
Lettenmeier, P. et al. Nanosized IrOx-Ir catalyst with relevant activity for anodes of proton exchange membrane electrolysis produced by a cost-effective procedure. Angew. Chem. Int. Ed. 55, 742–746 (2016).
Paoli, E. A. et al. Oxygen evolution on well-characterized mass-selected Ru and RuO2 nanoparticles. Chem. Sci. 6, 190–196 (2015).
Reier, T., Oezaslan, M. & Strasser, P. Electrocatalytic oxygen evolution reaction (OER) on Ru, Ir, and Pt catalysts: a comparative study of nanoparticles and bulk materials. ACS Catal. 2, 1765–1772 (2012).
Cherevko, S. et al. Dissolution of noble metals during oxygen evolution in acidic media. ChemCatChem 6, 2219–2223 (2014).
Reier, T. et al. Molecular insight in structure and activity of highly efficient, low-Ir Ir-Ni oxide catalysts for electrochemical water splitting (OER). J. Am. Chem. Soc. 137, 13031–13040 (2015).
Smith, R. D., Prevot, M. S., Fagan, R. D., Trudel, S. & Berlinguette, C. P. Water oxidation catalysis: electrocatalytic response to metal stoichiometry in amorphous metal oxide films containing iron, cobalt, and nickel. J. Am. Chem. Soc. 135, 11580–11586 (2013).
Lu, X. & Zhao, C. Electrodeposition of hierarchically structured three-dimensional nickel-iron electrodes for efficient oxygen evolution at high current densities. Nat. Commun. 6, 6616 (2015).
Shen, M. et al. Atomic scale analysis of the enhanced electro- and photo-catalytic activity in high-index faceted porous NiO nanowires. Sci. Rep. 5, 8557 (2015).
Sun, T. et al. Ordered mesoporous nickel sphere arrays for highly efficient electrocatalytic water oxidation. ACS Catal. 6, 1446–1450 (2016).
Vineesh, T. V. et al. Controllably alloyed, low density, free-standing Ni-Co and Ni-graphene sponges for electrocatalytic water splitting. Sci. Rep. 6, 31202 (2016).
Menezes, P. W. et al. Using nickel manganese oxide catalysts for efficient water oxidation. Chem. Commun. 51, 5005–5008 (2015).
Wang, S., Nai, J., Yang, S. & Guo, L. Synthesis of amorphous Ni−Zn double hydroxide nanocages with excellent electrocatalytic activity toward oxygen evolution reaction. ChemNanoMat 1, 324–330 (2015).
Klaus, S., Cai, Y., Louie, M. W., Trotochaud, L. & Bell, A. T. Effects of Fe electrolyte impurities on Ni(OH)2/NiOOH structure and oxygen evolution activity. J. Phy. Chem. C 119, 7243–7254 (2015).
Li, Y.-F. & Selloni, A. Mechanism and activity of water oxidation on selected surfaces of pure and Fe-doped NiOx . ACS Catal. 4, 1148–1153 (2014).
Liang, H. et al. Hydrothermal continuous flow synthesis and exfoliation of NiCo layered double hydroxide nanosheets for enhanced oxygen evolution catalysis. Nano Lett. 15, 1421–1427 (2015).
Jang, D. M. et al. Transition-metal doping of oxide nanocrystals for enhanced catalytic oxygen evolution. J. Phy. Chem. C 119, 1921–1927 (2015).
Burke, M. S. et al. Revised oxygen-evolution-reaction activity trends for first-row transition metal (oxy)hydroxides in alkaline media. J. Phys. Chem. Lett. 6, 3737–3742 (2015).
McCrory, C. C., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977–16987 (2013).
Huang, J. et al. CoOOH nanosheets with high mass activity for water oxidation. Angew. Chem. Int. Ed. 54, 8722–8727 (2015).
Tang, C., Cheng, N., Pu, Z., Xing, W. & Sun, X. NiSe nanowire film supported on nickel foam: an efficient and stable 3D bifunctional electrode for full water splitting. Angew. Chem. Int. Ed. 54, 9351–9355 (2015).
Ma, T., Dai, S., Jaroniec, M. & Qiao, S. Metal-organic framework derived hybrid Co3O4-carbon porous nanowire arrays as reversible oxygen evolution electrodes. J. Am. Chem. Soc. 136, 13925–13931 (2014).
Wang, H.-Y., Xiao, F.-X., Yu, L., Liu, B. & Lou, X. W. Hierarchical α-MnO2 nanowires@Ni1-xMnxOy nanoflakes core-shell nanostructures for supercapacitors. Small 10, 3181–3186 (2014).
Feng, J. X. et al. FeOOH/Co/FeOOH hybrid nanotube arrays as high-performance electrocatalysts for the oxygen evolution reaction. Angew. Chem. Int. Ed. 55, 3694–3698 (2016).
Chen, P. et al. Metallic Co4N porous nanowire arrays activated by surface oxidation as electrocatalysts for the oxygen evolution reaction. Angew. Chem. Int. Ed. 54, 14710–14714 (2015).
Li, S. et al. Co-Ni-based nanotubes/nanosheets as efficient water splitting electrocatalysts. Adv. Energy Mater. 6, 1501661 (2016).
Nai, J., Tian, Y., Guan, X. & Guo, L. Pearson’s principle inspired generalized strategy for the fabrication of metal hydroxide and oxide nanocages. J. Am. Chem. Soc. 135, 16082–16091 (2013).
He, K. et al. In situ decomposition of metal-organic frameworks into ultrathin nanosheets for the oxygen evolution reaction. Nano Res. 9, 1856–1865 (2016).
Ma, R., Liang, J., Takada, K. & Sasaki, T. Topochemical synthesis of Co−Fe layered double hydroxides at varied Fe/Co ratios unique intercalation of triiodide and its profound effect. J. Am. Chem. Soc. 133, 613–620 (2011).
McIntyre, N. S. & Zetaruk, D. G. X-ray photoelectron spectroscopic studies of iron oxides. Anal. Chem. 49, 1521–1529 (1977).
Long, X. et al. Metallic iron-nickel sulfide ultrathin nanosheets as a highly active electrocatalyst for hydrogen evolution reaction in acidic media. J. Am. Chem. Soc. 137, 11900–11903 (2015).
Enman, L. J., Burke, M. S., Batchellor, A. S. & Boettcher, S. W. Effects of intentionally incorporated metal cations on the oxygen evolution electrocatalytic activity of nickel (oxy)hydroxide in alkaline media. ACS Catal. 6, 2416–2423 (2016).
Sing, K. S. W. & Williams, R. T. Physisorption Hysteresis Loops and the Characterization of Nanoporous Materials. Adsorpt. Sci. Techno. 22, 773–782 (2004).
Lu, Z. et al. Superaerophobic electrodes for direct hydrazine fuel cells. Adv. Mater. 27, 2361–2366 (2015).
Diaz-Morales, O., Ledezma-Yanez, I., Koper, M. T. M. & Calle-Vallejo, F. Guidelines for the rational design of Ni-based double hydroxide electrocatalysts for the oxygen evolution reaction. ACS Catal. 5, 5380–5387 (2015).
Zhang, B. et al. Homogeneously dispersed multimetal oxygen-evolving catalysts. Science 352, 333–337 (2016).
Dong, C. et al. 3D binder-free Cu2O@Cu nanoneedle arrays for high-performance asymmetric supercapacitors. J. Mater. Chem. A. 2, 18229–18235 (2014).
Burke, M. S., Kast, M. G., Trotochaud, L. & Smith, A. M. & Boettcher, S. W. Cobalt-iron (oxy)hydroxide oxygen evolution electrocatalysts: the role of structure and composition on activity, stability, and mechanism. J. Am. Chem. Soc. 137, 3638–3648 (2015).
Sato, H. et al. Templating effects on the mineralization of layered inorganic compounds: (1) density functional calculations of the formation of single-layered magnesium hydroxide as a brucite model. Langmuir 19, 7120–7126 (2003).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77 (1996).
VandeVondele, J. et al. Quickstep: fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commu. 167, 103–128 (2005).
Hutter, J., Iannuzzi, M., Schiffmann, F. & VandeVondele, J. CP2K: atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 4, 15–25 (2014).
Jain, A. et al. Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 84 (2011).
Wang, L., Maxisch, T. & Ceder, G. Oxidation energies of transition metal oxides within the GGA + U framework. Phys. Rev. B 73 (2006).
VandeVondele, J. H. & H., J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 127, 114105–114109. (2007).
Goedecker, S., Teter, M. & Hutter, J. Separable Dual Space Gaussian Pseudo-potentials Gaussian Pseudopotentials. Phys. Rev. B 54 1703–1710 (1996).
Hartwigsen, C., Goedecker, S. & Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 58, 3641–3662 (1998).
Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 114, 145–152 (2005).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21402136, 21603161), Natural Science Foundation of Tianjin City (16JCYBJC17000), and the Specialized Research Fund for the Doctoral Program of Higher Education of China (20133201120004). Y.Z. acknowledges support from the “Talent Program” of Tianjin University of Technology and “Youth Thousand Talents Program” of Tianjin City. The authors acknowledge useful discussions with Prof. Yi Ding.
Author information
Authors and Affiliations
Contributions
Y.Z., Z.C., and T.Z. designed and conceived the experiments. T.Z. performed experiments and organized the data. T.Z., J.H., and Y.Z. wrote the manuscript. H.M., G.Z., H.W., and Y.L. were involved in discussion. J.H. and P.Z. performed DFT calculations. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhou, T., Cao, Z., Zhang, P. et al. Transition metal ions regulated oxygen evolution reaction performance of Ni-based hydroxides hierarchical nanoarrays. Sci Rep 7, 46154 (2017). https://doi.org/10.1038/srep46154
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46154
This article is cited by
-
Electrochemical Deposited Amorphous Bimetallic Nickle-Iron (Oxy)hydroxides Electrocatalysts for Highly Efficient Oxygen Evolution Reaction
Electrocatalysis (2023)
-
Manipulating Spin Polarization of Defected Co3O4 for Highly Efficient Electrocatalysis
Transactions of Tianjin University (2022)
-
Urea oxidation electrocatalysis on nickel hydroxide: the role of disorder
Journal of Solid State Electrochemistry (2021)
-
Partially amorphous NiFe-based bimetallic hydroxide nanocatalyst for efficient oxygen evolution reaction
Applied Physics A (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
