
Abstract
Colossoma macropomum, or tambaqui, is the largest native Characiform species found in the Amazon and Orinoco river basins, yet few resources for genetic studies and the genetic improvement of tambaqui exist. In this study, we identified a large number of single-nucleotide polymorphisms (SNPs) for tambaqui and constructed a high-resolution genetic linkage map from a full-sib family of 124 individuals and their parents using the genotyping by sequencing method. In all, 68,584 SNPs were initially identified using minimum minor allele frequency (MAF) of 5%. Filtering parameters were used to select high-quality markers for linkage analysis. We selected 7,734 SNPs for linkage mapping, resulting in 27 linkage groups with a minimum logarithm of odds (LOD) of 8 and maximum recombination fraction of 0.35. The final genetic map contains 7,192 successfully mapped markers that span a total of 2,811 cM, with an average marker interval of 0.39 cM. Comparative genomic analysis between tambaqui and zebrafish revealed variable levels of genomic conservation across the 27 linkage groups which allowed for functional SNP annotations. The large-scale SNP discovery obtained here, allowed us to build a high-density linkage map in tambaqui, which will be useful to enhance genetic studies that can be applied in breeding programs.
Similar content being viewed by others
Introduction
Tambaqui (Colossoma macropomum) is the most important native aquaculture species in Brazil. In 2014, production reached 139,209 tons1. This species has been broadly farmed in different Amazon boundary countries and has been introduced to other Latin American countries2, as it is well-suited to aquaculture farming, accepts artificial feed, grows rapidly, and is accepted by consumer markets3. However, genetic studies of tambaqui are limited to genetic diversity surveys4,5,6, transcriptome analysis under specific conditions7, some SNPs markers8, and mitochondrial genome sequencing9.
Genetic linkage maps are essential resources for genomic and genetic research. They provide frameworks for understanding genome structure and function. Linkage maps are essential for quantitative trait locus (QTL) identification and mapping. QTL mapping is an important way to identify genes that are related to trait variations within and between populations or species, allowing for implementation of genetic and breeding programs10. Genetic maps can be used to facilitate genome assembly corrections, anchoring scaffolds onto linkage groups to build chromosomal assembly11. With interspecific crosses, high-density genetic mapping can provide a genome-scan of segregation distortion within the genome12 and can investigate genomic incompatibilities between species at the genome level13. Genetic studies such as genetic linkage mapping14, QTL mapping15, populational genetic analysis16, and genome-wide association studies17 require a large number of reliable molecular markers across the genome.
Single nucleotide polymorphisms (SNPs) are the marker of choice for genetic studies in various organisms. SNPs are the most abundant molecular markers in any vertebrate genome, with a SNP present in 100–500 bp on average18. SNPs are mostly bi-allelic, making them amenable for high-throughput genotyping using SNP arrays19,20. Over the last decade, with the development of next-generation sequencing technologies21,22,23, genome-scale SNP markers can now be efficiently and cost-effectively identified in any organism for which prior genomic information does not yet exist24. Genotype-by-sequencing (GBS) is one next-generation sequencing technique, and is based on the reduction of genome complexity using restriction enzymes23. GBS is characterized as a simple, quick, specific, reproducible technique23, and has been extensively used to identify a large number of SNPs in various model and non-model species25,26,27,28.
In the present study, we applied GBS to identify large-scale SNPs and construct a high-density genetic linkage map for tambaqui, using the Illumina HiSeq 2500 platform. In addition, we conducted syntenic relationship and functional annotation analyses by aligning tambaqui against the zebrafish (Danio rerio) genome. This study provides large-scale SNP markers and high-density linkage maps in tambaqui, which can be a useful resource for facilitating the tambaqui physical map construction, genome assembly, and QTL mapping to enhance genetic studies and breeding programs.
Results
Enzyme selection
Based on in silico and in vitro genomic fragmentation, we tested whether the enzymes PstI and SbfI would generate the expected number of reads required to obtain ~7X sequencing coverage29. Each enzyme generated a different distribution of fragment lengths across the entire genome (Supplementary Figure S1). The enzyme PstI yielded a larger number of fragments that ranged between 200 bp and 500 bp (see Supplementary Figure S1), providing suitable sizes for GBS to be clustered on cBOT (Illumina) in bridge amplification.
Sequencing
As summarized in Table 1, the 126 samples generated over 352 million single-end reads that had a length of 100 bp. After read trimming, ~285 million quality reads (81%) were obtained, with over 27 Giga bases, equivalent to >8X genome coverage. An average of 3.2 million quality reads were obtained from the parents and over 2.2 million quality reads were obtained from each progeny (Table 1). The vast majority of samples had genome sequencing depth >8X, and only 15 progenies had fewer than one million reads (~4.5X) (Supplementary Spreadsheet S1).
SNP discovery
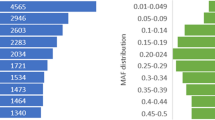
A total of 81,222 pairwise alignments were obtained with the UNEAK pipeline. After filtering with default parameters, 68,584 putative SNPs that had a minor allele frequency (MAF) that was greater than 0.05 (Table 2) were identified. SNPs were classified into transitions (Ti) and transversions (Tv) based on nucleotide substitution. The number of A/G transitions was about two times greater than C/T transitions; the numbers of A/C and A/T transversions were relatively higher than the C/G and G/T transversions. Transitions are the most common type of nucleotide substitutions, and in this sample 64% of the base changes were transitions and 36% were transversions, with an observed transition to transversion ratio (Ti/Tv) of 1.8:1.
SNP filtering
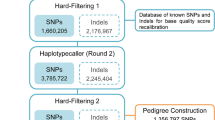
A set of 10,288 high quality SNP markers that had a sample call rate >80% and that were heterozygous for at least one parent were retained for further analysis. After removing markers with significant segregation distortion, a total of 7,734 SNPs were retained for genetic map construction. Of these SNPs, 3,641 were heterozygous only in female, 2,565 were heterozygous only in male, and the remaining 1,528 were heterozygous in both parents. A total of 118 samples with a SNP call rate >80% were retained for genetic map construction.
Construction of the high-density linkage map
A total of 7,192 SNPs were mapped to 27 linkage groups, consistent with tambaqui’s haploid chromosome number (n = 27)30 (Supplementary Spreadsheet S2). The genetic map spanned a total of 2,810.9 cM, with an average marker-interval of 0.39 cM (Fig. 1). On average, each linkage group contained 266 markers that spanned an average length of 104 cM, with an average marker interval of 0.41 cM. The number of mapped markers per linkage group varied from 86 markers on LG14 to 362 markers on LG2 and LG9. The smallest linkage group was LG25, which contained 120 markers spanning a length of 70.94 cM. The largest linkage group was LG2, which had 362 markers and a length of 145.66 cM. The maximum gap size in each linkage group ranged from 2.08 cM on LG20 to 14.79 cM on LG12, with an average of 5.27 cM (Table 3).

The 27 linkage groups are represented by vertical bars while the horizontal lines represent markers. Genetic distances between adjacent markers are shown on the side ruler in centiMorgan (cM).
The distribution of markers across each linkage group was assessed using the sliding window approach. The number of markers within a window was counted using a sliding window of 10 cM with a step size of 1 cM. The density value for each window was calculated by dividing the total of markers within a window by the window length. As shown in Fig. 2, the SNP markers are evenly distributed across the 27 linkage groups. The linkage groups LG2, LG4, LG6, LG8, LG9, LG12, and LG22 had windows with high density of markers (>8 markers per cM), most of which were clustered at the same genetic positions. LG14 had more windows with low density of markers. LG12 had the window with the highest density (10.4 markers per cM) and also the window with the lowest density (0 markers per cM). In general, the regions with high marker density were located near the centromeres (Fig. 2).

The dotted box indicates regions with higher marker density. The red lines indicate gaps without markers.
Comparative genomic analysis
Out of the 7,192 SNP markers mapped to the linkage map, 1,237 markers that contained sequences were successfully aligned against the zebrafish genome (Fig. 3). The synteny analysis between tambaqui and zebrafish showed variable levels of genomic conservation across the 27 linkage groups. Most tambaqui linkage groups showed a relationship with homologous zebrafish chromosomes. Through comparative analysis, a high level of genomic conservation was found between tambaqui and zebrafish for most linkage groups. For instance, more than 30% of the SNPs mapped in LGs 17, 3, 19, 14, 10, 18, 1, 11, 8, 25, 21, 6, 20, and 15 of tambaqui were aligned on zebrafish chromosomes 21, 15, 23, 7, 8, 3, 9, 5, 13, 1, 10, 6, 17, and 18, respectively, suggesting orthologous chromosomal relationships. Zebrafish chromosome 7 corresponded to tambaqui LGs 14 and 16, while zebrafish chromosome 5 corresponded to LGs 11 and 23 in tambaqui. LG 27 in tambaqui had a similar level of consensus between homologous chromosomes 11 and 22 in zebrafish. However, LGs 2, 4, 5, 13, 22, 24, and 26 in tambaqui appear scattered among several chromosomes, suggesting large-scale chromosomal rearrangements between species.

Syntenic links between tambaqui linkage map and zebrafish genome were made with the Circos software (A). Plot of the percentage of SNPs that are shared between tambaqui linkage groups and zebrafish chromosomes (B). In synteny comparison, the connecting links are color coded according to the zebrafish chromosomes arc to their tambaqui linkage-group corresponding.
Functional annotation
The SNPs for each linkage group were annotated against the genes from the zebrafish (ENSEMBL release 84). This annotation allowed us to evaluate the potential use of our genetic map with respect to possible important traits in breeding programs. Approximately 60% (742) of the SNPs that were evaluated were annotated to be intronic (36%), downstream (13%), or upstream (11%) of gene regions. These SNPs could thus have a direct effect on or be associated with potential performance traits of interest (Table 4). The other 40% were evaluated as synonymous variant (11%), intergenic variant (10%), missense variant (7%), non-coding transcript variant (4%), splice region variant (3%), 3 prime UTR variant (1%) and other variants (4%). Only 2% of the SNPs resulted in a stop lost codon, and 1% results in a stop gain codon, both of which can lead to protein truncation (Supplementary Spreadsheet S3).
Allele frequency and coverage evaluation
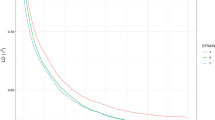
To evaluate the GBS approach’s capability in determining the accuracy of allelic frequencies, we compared the allelic frequency obtained in our genotyping with the frequency expected for a diploid cross. We also analyzed allele frequency calls into population using different SNP coverage thresholds (Fig. 4). The SNPs were placed into six groups, depending on coverage (5–10X, 10–20X, 20–40X, 40–80X, 80–160X, ≥160X). For SNPs with coverage ≥160X, we observed three peaks in allele frequency distribution. The first and third peak represent the crosses of AA x Aa, with allele frequencies of 0.25 and 0.75, respectively, with a 1:1 segregation of the AA and Aa genotypes. The second peak represents the cross Aa x Aa, with allele frequencies of 0.5 and a 1:2:1 segregation of the AA, Aa, and aa genotypes. Lower coverage resulted in allelic frequency deviations from expected frequencies.

Effect of coverage depth on allele frequency of tambaqui (Colossoma macropomum) genotyped with GBS.
Discussion
Developing molecular marker panels and genetic linkage maps are essential for genetic improvement programs in aquaculture. Despite the economic and ecological importance of tambaqui, limited genomic resources are available. In this study, we report the first genome-scale SNP discovery and high-density genetic map construction in tambaqui. Sequencing the family under study allowed for the identification and genotyping of a large number of markers in an efficient and cost-effective way. The linkage mapping analysis resulted in the same number of linkage groups as corresponds to tambaqui’s haploid chromosome number (n = 27)30. In addition, this process created a high-density linkage map.
SNP discovery and genetic map construction using the GBS approach have been conducted in a number of aquaculture species including Asian seabass (Lates calcarifer)31, common pandora (Pagellus erythrinus)32, sablefish (Anoplopoma fimbria)33, scallop (Chlamys farreri)34, oysters (Crassostrea gigas × Crassostrea angulata)34, and small abalone (Haliotis diversicolor)13,14,24,31,32,34,35. This tambaqui genetic map will be valuable for genomic research and genetic enhancement applications, such as identifying interspecific hybrids30 and implementing genome-wide association studies36.
Successfully applying the GBS approach depends on choosing an effective restriction enzyme. This choice determines the number of genomic fragments produced by the complexity reduction for the species under study, which in turn are used to identify genome-wide sequence polymorphisms37. The in silico fragmentation showed that PstI and SbfI produce a large difference in the number of restriction sites. The PstI enzyme was more suitable for the fragmentation of the genome tested. This enzyme has also been successfully used in prawn kuruma (Marsupenaeus japonicas) for high-resolution genetic linkage map construction and QTL mapping35.
The per-base quality of sequencing reads has a great impact on the accuracy of marker detection and genotype calling38. In our analysis, we trimmed off 19% of all reads with low-quality scores. This decision resulted in 28% larger numbers of identified SNPs than that from non-trimed reads (unpublished data). This is because the UNEAK Pipeline performs SNP discovery based on a network of SNPs formed by reads that have only a base incompatibility. Low-quality reads create more complicated networks during SNP discovery; it also reduces read counts below a specified error tolerance rate of 0.03. The UNEAK Pipeline considers this networks as a result of sequencing errors and exclude them from genotyping39, reducing SNP coverage. In our analysis, SNP coverage had a direct effect on allelic frequency (Fig. 4). The results showed that higher SNP coverage results in an allele frequency distribution that is consistent with an expected diploid crossing. For instance, the coverage of ≥160X provided smaller levels of missing data and a higher percentage of called genotypes (Fig. 4).
GBS was an efficient method for discovering tens of thousands of SNPs in a genome that had no reference sequence. However, genetic maps require high-quality genotypes of markers from a certain number of mapping samples. In this study, we used high criteria to SNP call rate and sample call rate. Although the filtering steps eliminated 85% of the 68,584 SNPs, the remaining markers and samples had high-quality scores that ensured the construction of genetic map with a high level of accuracy. Similar numbers of makers were also reported in other studies using GBS strategies31,34,40,41, which generally show high levels of missing genotypes due to the relatively low sequencing depth.
The GBS approach generated marker-containing sequences, allowing for analysis with other closely-related model species based on sequence homology (Fig. 3). About 17% of the tambaqui SNPs were successfully aligned with the zebrafish genome. This percentage is higher than that reported in Mexican tetra (Astyanax mexicanus) (14.2%)11. However, this higher alignment could be a consequence of the genetic distance between the species used or a consequence of manually maximizing the Bowtie alignment parameters, since that our alignment, using the default parameter, falls from 17% to 9%. A variable degree of synteny is observed between tambaqui linkage groups and zebrafish chromosomes, with some linkages groups showing homology with several chromosomes of zebrafish. The majority of the linkage groups in tambaqui have one-to-one orthologous relationships with chromosomes zebrafish. Notably, zebrafish chromosomes 5 and 7 have two-to-one syntenic relationships with tambaqui linkage groups. The use of zebrafish to perform synteny and collinearity analyses may provide a framework for mapping candidate genes responsible for the traits related to phenotypic divergence in tambaqui. Zebrafish genome also enables the transfer of genomic information between zebrafish and others species, such as Mexican tetra11, gudgeons42, common carp43, rainbow trout44, and channel catfish45.
The syntenic relationships and functional annotation of zebrafish allowed for annotation of the SNPs from the tambaqui linkage map. In turn, this allowed for the identification of 1,237 variants, from which 36% were annotated in genomic regions (Supplementary Spreadsheet S3). Candidate genes for important traits in tambaqui were identified, such as insulin-like growth factor binding protein (IGFBP), a hypoxia-inducible gene that acts in regulating embryonic growth and development under hypoxic stress46. We also found variants in genes that are of medical interest, such as insulin-like growth factor 1a receptor (IGF1RA), a gene that has been studied in animals with relatively slow progression for insulin resistance to better understand possible genes or metabolic situations that may accelerate diabetes progression, thereby offering possible new therapeutic targets47. In addition, variants of upstream, downstream, and intronic regions of the immunoglobulin heavy chain variable (IGHV) gene were annotated. Interesting, this gene has been reported to be the most important indicator of chronic lymphocytic leukemia prognosis48. Although the tambaqui genome sequences are still not available, comparative genome analysis enabled the identification of large number of markers in genic regions, which in turn could be used in tambaqui breeding programs.
Conclusion
This study demonstrated that GBS is an effective approach for SNP discovery and the development of high-density genetic linkage maps in tambaqui. This study was also the first to identify genome-scale SNP markers and to construct high-density genetic maps for tambaqui. Comparative genomic analysis with zebrafish revealed variable levels of homologous relationships with zebrafish chromosomes for all tambaqui linkage groups. In addition, large numbers of markers in genic regions were annotated, and genes with potential functions for performance traits were identified. The SNPs and genetic map reported in this work should be valuable tools for genetic studies and aquaculture improvements in tambaqui.
Materials and Methods
Ethical statement
All experimental protocols employed in the present study that relate to animal experimentation were performed in accordance with Brazilian Directive for the Care and Use of Animals in Teaching or Scientific Research Activities, resolution number 30/2016 approved by the National Animal Experimentation Control Council to ensure compliance with international guidelines for animal welfare.
The samples were acquired from a commercial hatchery and were not subjected to any experimental manipulation or euthanasia. No specific permits were required for the work described here.
Fish materials and DNA extraction
The Colossoma macropomum subjects used in this study were collected at a fisheries farm located in the Rondônia state, in the northwestern region of Brazil. A full-sib F1 family of 124 offspring was created by crossing a wild female with a wild male. When the subject’s mean body weight reached ~6 g (mean body length of ~5.5 cM), tail fin clips were collected from each progeny and the two parents, and preserved in 90% ethanol. DNA extraction followed these steps: proteinase K digestion (Promega), DNA precipitation in absolute ethanol, washing in 70% ethanol, and resuspension in ultrapure water. DNA concentration was quantified using a Qubit 2.0 fluorimeter (Invitrogen, Carlsbad, CA, USA) and Nanodrop®2000c spectrophotometer. DNA integrity was checked in 1% agarose gel. All DNA samples were stored at −20 °C prior to sequencing library preparation.
Construction of GBS sequencing libraries
To select an enzyme that uniformly distributed cutting sites across the tambaqui genome, we performed an in-silico DNA cleavage on the zebrafish genome with PstI and SbfI in R using the subsequent Bioconductor49 packages: Biostrings, BSgenome.Drerio.UCSC.danRer7, plyr, ggplot2, reshape2 and scales (https://github.com/). Additionally, we performed in vitro genomic cleavage of tambaqui DNA samples with the PstI and SbfI enzymes, according to the manufacturer’s protocol (New England BioLabs®).
GBS library construction and sequencing were conducted at the Animal Biotechnology Laboratory at the University of São Paulo (Piracicaba, Brazil), using the protocol described by Elshire et al.23 with modifications. In brief, 100 ng of high-quality DNA were cleaved with 0.2 μL PstI (10U/uL) at 37 °C for 2 hours. After digestion, the restriction enzyme was deactivated at 85 °C for 20 seconds, and the samples were dehydrated. To perform the ligation reaction, all samples were rehydrated in 6 μL of adapter solutions and incubated at 22 °C for 2 hours in binding mix with T4 DNA ligase (New England BioLabs®). For post-ligation reactions, 10 μl from each of the 126 samples (124 progenies + 2 parents) were aliquoted and pooled with 32 samples per group, followed by polymerase chain reaction (PCR) cleanup using the QIAquick PCR Purification Kit® (Quiagen), resulting in four “pre-PCR” GBS libraries. Within each library, PCR amplification was conducted using specific primers for sequencing, using the Illumina platform. PCR purification was performed using the Agencourt AMPure XP PCR purification kit®. PCR products were quantified by quantitative PCR, using the KAPA Library Quantification Kit (KAPA Biosystems). For each library, 2 pools of 32 samples were mixed and diluted to 16 pM. For each library, 64 barcoded samples were pooled. The libraries were clustered using TruSeq SR Cluster Kit v3-cBot-HS on the cBOT (Illumina) equipment. The libraries were sequenced using Illumina TruSeq SBS Kit v3-HS on the Illumina HiSeq2500 sequencer (Illumina, San Diego) on two lanes of single-end reads with a read length of 100 bp.
Read processing and SNP discovery
For all samples, quality trimming was performed with SeqyClean tool v. 1.9.10 (https://bitbucket.org/izhbannikov/seqyclean/) using Phred quality score ≥ 24 and fragment size ≥50. A contaminant database was provided to the program to remove vector, adapter, and other sequence contaminations. The sequence processing was performed using the UNEAK39 (Universal Network Enabled Analysis Kit) pipeline with default parameters. UNEAK separates all reads that have an exact match to a barcode plus the subsequent five nucleotides that are expected to remain from a PstI cut-site (i.e., 5′… CTGCA’G…3′) but no missing data in the first 64 bp subsequently the barcode. Identical reads were clustered into tags; rare or singleton tags represented by fewer than five reads were excluded to reduce possible sequencing errors. All tags were then aligned pair wisely, and 1-bp mismatches were detected as potential SNPs. To reduce false SNP calls, the UNEAK pipeline applied an error tolerance filter of 0.03.
Linkage map construction
The UNEAK output files were imported to Tassel v.3.050 in order to apply post-UNEAK filtering. Genotype data were filtered for both parent and progeny samples. Only markers that had no missing genotypes for both parents were retained. Were applied a sample call rate and marker call rate of >80% (i.e., at least 80% SNPs had genotypes called in each sample and a SNP was called in at least 80% of the samples). In addition, only markers that were heterozygous in at least one of the two parents (i.e., AA x AB, AB x AA, or AB x AB) were used for linkage mapping analysis. Next, markers that significantly deviated from Mendelian inheritance, as determined by a chi-squared test (P < 0.001), were excluded. The genetic maps were constructed using the pseudo-testcross strategy. The program R/OneMap was used to assign markers to linkage groups that had a minimum logarithm of odds (LOD) score of 8 and a maximum recombination fraction of 0.35. The program JoinMap4 was then used to construct the map for each linkage group, using the regression mapping algorithm and Kosambi mapping function.
Analysis of syntenic relationships
The SNPs in the tambaqui linkage maps were used to analyze the syntenic relationships with zebrafish (Danio rerio). The sequence reads that harbored the mapped SNPs were extracted and aligned with the zebrafish genome (Danio rerio, Zv9) using Bowtie2 v.2.2.5. To maximize the number of alignments, the parameters -D, -R, -N, -L, and -i were optimized manually (-D 100 -R 7 -N 1 -L 20 -i S,1,0.01). Sequences with multiple alignments were removed. The Circos51 software was used to plot the relationship between the zebrafish chromosomes and tambaqui linkage groups.
Functional annotation
The SNPs that successfully aligned with the zebrafish genome (Danio rerio, Zv9) were annotated using the Variant Effect Predictor (VEP) tool v.7152. The annotations included a range of variant types such as intronic, downstream gene, upstream gene, synonymous, missense, intergenic, splice region, and non-coding transcript.
Additional Information
How to cite this article: Nunes, J. R. S. et al. Large-scale SNP discovery and construction of a high-density genetic map of Colossoma macropomum through genotyping-by-sequencing. Sci. Rep. 7, 46112; doi: 10.1038/srep46112 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
IBGE. Produção da Pecuária Municipal 2014. Instituto Brasileiro de Geografia e Estatística 42, 1–39 (2014).
Kapetsky, J. M. & Nath, S. S. A strategic assessment of the potential for freshwater fish farming in Latin America: Annex 2, Water Temperature Model. COPESCAL Technical Paper 10, (Food and Agriculture Organization of the United Nations, 1997).
Lima, C., de S., Bomfim, M. A. D., Siqueira, J. C. de, Ribeiro, F. B. & Lanna, E. A. T. Crude protein levels in the diets of tambaqui, Colossoma macropomum (Cuvier, 1818), fingerlings. Rev. Caatinga 29, 183–190 (2016).
Santos, M. C. F., Ruffino, M. L. & Farias, I. P. High levels of genetic variability and panmixia of the tambaqui Colossoma macropomum (Cuvier, 1816) in the main channel of the Amazon River. J. Fish Biol. 71, 33–44 (2007).
Jacometo, C. B. et al. Variabilidade genética em tambaquis (Teleostei: Characidae) de diferentes regiões do Brasil. Pesqui. Agropecuária Bras. 45, 481–487 (2010).
Santos, C. H. A., Santana, G. X., Sá Leitão, C. S., Paula-Silva, M. N. & Almeida-Val, V. M. F. Loss of genetic diversity in farmed populations of Colossoma macropomum estimated by microsatellites. Anim. Genet. 47, 373–376 (2016).
Prado-Lima, M. et al. Transcriptomic Characterization of Tambaqui (Colossoma macropomum, Cuvier, 1818) Exposed to Three Climate Change Scenarios. PLoS One 11, e0152366 (2016).
Martínez, J. G. et al. SNPs markers for the heavily overfished tambaqui Colossoma macropomum, a Neotropical fish, using next-generation sequencing-based de novo genotyping. Conserv. Genet. Resour. online, 1–5 (2016).
Wu, Y.-P., Xie, J.-F., He, Q.-S. & Xie, J.-L. The complete mitochondrial genome sequence of Colossoma macropomum (Characiformes: Serrasalmidae). Mitochondrial DNA 1–2 doi: 10.3109/19401736.2014.1003853 (2015).
Ding, G. et al. Quantitative trait loci for seed yield and yield-related traits, and their responses to reduced phosphorus supply in Brassica napus. Ann. Bot. 109, 747–759 (2012).
Carlson, B. M., Onusko, S. W. & Gross, J. B. A High-Density Linkage Map for Astyanax mexicanus Using Genotyping-by-Sequencing Technology. G3&#58; Genes|Genomes|Genetics 5, 241–251 (2015).
Bodénès, C., Chancerel, E., Ehrenmann, F., Kremer, A. & Plomion, C. High-density linkage mapping and distribution of segregation distortion regions in the oak genome. DNA Res. 23, 115–124 (2016).
Liu, S. et al. High-density interspecific genetic linkage mapping provides insights into genomic incompatibility between channel catfish and blue catfish. Anim. Genet. 47, 81–90 (2016).
Ren, P. et al. Genetic mapping and quantitative trait loci analysis of growth-related traits in the small abalone Haliotis diversicolor using restriction-site-associated DNA sequencing. Aquaculture 454, 163–170 (2016).
Yu, H. et al. Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 6, e17595 (2011).
Yáñez, J. M. et al. Genome-wide single nucleotide polymorphism (SNP) discovery in Atlantic salmon (Salmo salar): validation in wild and farmed American and European populations. Mol. Ecol. Resour. n/a-n/a, doi: 10.1111/1755-0998.12503 (2016).
Correa, K. et al. Genome wide association study for resistance to Caligus rogercresseyi in Atlantic salmon (Salmo salar L.) using a 50K SNP genotyping array. Aquaculture doi: 10.1016/j.aquaculture.2016.04.008 (2016).
Vignal, A., Milan, D., SanCristobal, M. & Eggen, A. A review on SNP and other types of molecular markers and their use in animal genetics. Genet. Sel. Evol. 34, 275 (2002).
Liu, S. et al. Generation of genome-scale gene-associated SNPs in catfish for the construction of a high-density SNP array. BMC Genomics 12, 53 (2011).
Liu, S. et al. Development of the catfish 250K SNP array for genome-wide association studies. BMC Res. Notes 7, 135 (2014).
Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A. & Johnson, E. A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 17, 240–248 (2007).
Van Tassell, C. P. et al. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat. Methods 5, 247–252 (2008).
Elshire, R. J. et al. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS One 6, e19379 (2011).
Starks, H. A., Clemento, A. J. & Garza, J. C. Discovery and characterization of single nucleotide polymorphisms in coho salmon, Oncorhynchus kisutch. Mol. Ecol. Resour. 16, 277–287 (2016).
Poland, J. A., Brown, P. J., Sorrells, M. E. & Jannink, J. L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 7, e32253 (2012).
Annicchiarico, P. et al. Assessment of Cultivar Distinctness in Alfalfa: A Comparison of Genotyping-by-Sequencing, Simple-Sequence Repeat Marker, and Morphophysiological Observations. Plant Genome 9, 1–12 (2016).
Pértille, F. et al. High-throughput and Cost-effective Chicken Genotyping Using Next-Generation Sequencing. Sci. Rep. 6, 26929 (2016).
Boutet, G. et al. SNP discovery and genetic mapping using genotyping by sequencing of whole genome genomic DNA from a pea RIL population. BMC Genomics 17, 121 (2016).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PloS one 7, e37135 (2012).
Hashimoto, D. T., Senhorini, J. A., Foresti, F., Martínez, P. & Porto-Foresti, F. Genetic Identification of F1 and Post-F1 Serrasalmid Juvenile Hybrids in Brazilian Aquaculture. PLoS One 9, e89902 (2014).
Wang, L. et al. Construction of a high-density linkage map and fine mapping of QTL for growth in Asian seabass. Sci. Rep. 5, 16358 (2015).
Manousaki, T. et al. Exploring a Non-model Teleost Genome Through RAD Sequencing - Linkage Mapping in Common Pandora, Pagellus erythrinus and Comparative Genomic Analysis. G3 Genes Genomes Genet. 6, g3. 115.023432, (2015).
Rondeau, E. B. et al. Genomics of sablefish (Anoplopoma fimbria): expressed genes, mitochondrial phylogeny, linkage map and identification of a putative sex gene. BMC Genomics 14, 452 (2013).
Jiao, W. et al. High-Resolution Linkage and Quantitative Trait Locus Mapping Aided by Genome Survey Sequencing: Building Up An Integrative Genomic Framework for a Bivalve Mollusc. DNA Res. 21, 85–101 (2014).
Wang, J., Li, L. & Zhang, G. A High-Density SNP Genetic Linkage Map and QTL Analysis of Growth-Related Traits in a Hybrid Family of Oysters (Crassostrea gigas × Crassostrea angulata) Using Genotyping-by-Sequencing. G3 Genes|Genomes|Genetics 6, 1417–1426 (2016).
Lu, X. et al. High-resolution genetic linkage mapping, high-temperature tolerance and growth-related quantitative trait locus (QTL) identification in Marsupenaeus japonicus. Mol. Genet. Genomics 291, 1391–1405 (2016).
Perazza, C. A., de Menezes, J. T. B., Ferraz, J. B. S. & Hilsdorf, A. W. S. Lack of intermuscular bones in specimens of Colossoma macropomum: An unusual phenotype to be incorporated into genetic improvement programs. Aquaculture, doi: 10.1016/j.aquaculture.2016.05.014 (2016).
Poland, J. A. & Rife, T. W. Genotyping-by-Sequencing for Plant Breeding and Genetics. Plant Genome J. 5, 92–102 (2012).
Nielsen, R., Paul, J. S., Albrechtsen, A. & Song, Y. S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 12, 443–51 (2011).
Lu, F. et al. Switchgrass Genomic Diversity, Ploidy, and Evolution: Novel Insights from a Network-Based SNP Discovery Protocol. PLoS Genet. 9, e1003215 (2013).
Ward, J. A. et al. Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genomics 14, 2 (2013).
Li, C. et al. SNP discovery in wild and domesticated populations of blue catfish, Ictalurus furcatus, using genotyping-by-sequencing and subsequent SNP validation. Mol. Ecol. Resour. 14, 1261–1270 (2014).
Kakioka, R., Kokita, T., Kumada, H., Watanabe, K. & Okuda, N. A RAD-based linkage map and comparative genomics in the gudgeons (genus Gnathopogon, Cyprinidae). BMC Genomics 14, 32 (2013).
Zhang, X. et al. A Consensus Linkage Map Provides Insights on Genome Character and Evolution in Common Carp (Cyprinus carpio L.). Mar. Biotechnol. 15, 275–312 (2013).
Guyomard, R., Boussaha, M., Krieg, F., Hervet, C. & Quillet, E. A synthetic rainbow trout linkage map provides new insights into the salmonid whole genome duplication and the conservation of synteny among teleosts. BMC Genet. 13, 15 (2012).
Zhang, Y. et al. Comparative genomic analysis of catfish linkage group 8 reveals two homologous chromosomes in zebrafish and other teleosts with extensive inter-chromosomal rearrangements. BMC Genomics 14, 387 (2013).
Kajimura, S., Aida, K. & Duan, C. Understanding Hypoxia-Induced Gene Expression in Early Development: In Vitro and In Vivo Analysis of Hypoxia-Inducible Factor 1-Regulated Zebra Fish Insulin-Like Growth Factor Binding Protein 1 Gene Expression. Mol. Cell. Biol. 26, 1142–1155 (2006).
Maddison, L. a, Joest, K. E., Kammeyer, R. M. & Chen, W. Skeletal muscle insulin resistance in zebrafish induces alterations in β-cell number and glucose tolerance in an age- and diet-dependent manner. Am. J. Physiol. - Endocrinol. Metab. 308, E662–E669 (2015).
Ghia, P. et al. ERIC recommendations on IGHV gene mutational status analysis in chronic lymphocytic leukemia. Leukemia 21, 1–3 (2007).
Huber, W. et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015).
Glaubitz, J. C. et al. TASSEL-GBS: A High Capacity Genotyping by Sequencing Analysis Pipeline. PLoS One 9, e90346 (2014).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010).
Acknowledgements
We would like to thank the Amazon Research Foundation (FAPEAM – Process:1270/2012), CAPES (Pro-Amazônia), and São Paulo Research Foundation (FAPESP 15/23.883-0) for granting funds to support this project and FAPEAM and CAPES-PDSE for funding José de Ribamar da Silva Nunes’s scholarship. In addition, we are deeply indebted to Drs Sonia Andrade (University of São Paulo) for the discussion on GBS analysis and Dale Simpson and Ramona Davis for their help. LLC is a recipient of a CNPq productivity scholarship.
Author information
Authors and Affiliations
Contributions
J.R.S.N. analyzed the data. J.R.S.N. and S.L. wrote the manuscript. S.L. contributed to the data analysis and manuscript revision. F.P. helped with bioinformatics analysis and GBS implementation. P.M.S.V. helped with G.B.S. implementation. A.W.S.H. helped in design of the project. C.A.P. and A.W.S.H. contributed to the fish family maintenance and sample collection. V.M.F.A.V. and A.W.S.H. provided sequencing resources and reviewed the manuscript. Z.L. supervised data analysis and reviewed the manuscript. L.L.C. supervised the entire project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Nunes, J., Liu, S., Pértille, F. et al. Large-scale SNP discovery and construction of a high-density genetic map of Colossoma macropomum through genotyping-by-sequencing. Sci Rep 7, 46112 (2017). https://doi.org/10.1038/srep46112
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46112
This article is cited by
-
Continent-island boundary and environment-shaped evolution in the marine amphipod Ampithoe marcuzzii complex (Crustacea: Eumalacostraca: Ampithoidae)
Scientific Reports (2024)
-
A high-density linkage map and sex-linked markers for the Amazon Tambaqui Colossoma macropomum
BMC Genomics (2021)
-
Development of a multi-species SNP array for serrasalmid fish Colossoma macropomum and Piaractus mesopotamicus
Scientific Reports (2021)
-
Development of a SNP linkage map and genome-wide association study for resistance to Aeromonas hydrophila in pacu (Piaractus mesopotamicus)
BMC Genomics (2020)
-
A first genetic linage map construction and QTL mapping for growth traits in Larimichthys polyactis
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
