
Abstract
Using quantum mechanics calculations, we theoretically study the dissociation and ionization dynamics of the hydrogen-molecule ion in strong laser fields. Having prepared the nuclear wave packet of H2+ in a specific vibrational state, a pump laser is used to produce a vibrational excitation, leading to quasi-periodical vibration without ionization. Then, a time-delayed few-cycle laser is applied to trigger the dissociation or ionization of H2+. Both the time delay and the intensity of the probe laser alter the competition between dissociation and ionization. We also explore the dependence of kinetic-energy release spectra of fragments on the time delay, showing that the channels of above-threshold dissociation and below-threshold dissociation are opened and closed periodically. Also, dissociation from different channels is influenced by nuclear motion. The dissociation mechanism has been described in detail using the Floquet picture. This work provides a useful method for steering the electronic and nuclear dynamics of diatomic molecules in intense laser fields.
Similar content being viewed by others


Introduction
The irradiation of diatomic molecules by an intense laser pulse can yield very complex dynamics. In past decades, much attention has been given to relevant processes and phenomena in strong-field physics, such as charge-resonance-enhanced ionization (CREI)1,2, high-order harmonic generation (HHG)3,4, bond softening (BS)5, bond hardening (BH)6, above-threshold dissociation (ATD)7, and below-threshold dissociation (BTD)8. Ionization is a fundamental process in atoms and molecules in intense laser fields, and its probability can be calculated within Ammosov-Delone-Krainov theory based on the strong-field approximation9,10, within time-dependent density-functional theory11, or by solving the time-dependent Schrödinger equation (TDSE)12. The ionization dynamics of even the simplest molecule system, H2+, is more complicated than that of single atoms because of the additional vibrational dimension of the nuclei and the two-center effect. A molecular system undergoes multiple bursts of ionization within half cycles during a laser-field oscillation, in contrast to the widely accepted “tunnel ionization” picture of atoms13. Also, double-slit-like interferences have been observed in photo-ionization experiments14 on H2+ and explained theoretically15,16. In CREI, the ionization rate is maximized for large internuclear distance, exceeding the atom limit by an order of magnitude1,2,17,18,19,20. Litvinyuk and coworkers21 claimed to have first observed, experimentally, the elusive double-peak structure in the R-dependent ionization rate of H2+. The rapidity and large spatial distribution of nuclear wavepackets were the main reasons previous experiments failed to observe the double-peak structure. Based on the one-dimensional (1D) Coulomb potential, Qu et al. claimed that, theoretically, the ionization and HHG of H2+ in short intense laser pulses with moving nuclei are substantially different from those in the absence of nuclear correlation22. Using the similar 1D model in the non-Born-Oppenheimer (NBO) approach, Nguyen et al.23 found that the ionization probability initially increases and subsequently decreases as the vibrational level of H2+ increases.
Another important process in photochemical reactions is molecular dissociation. Extensive studies have been done on H2+, the simplest diatomic system. If the laser field is intense enough to break the chemical bond, H2+ displays two fragmentation channels: (i) the ionization channel, i.e., Coulomb explosion (CE), , and (ii) the dissociation channel,
, and (ii) the dissociation channel,  . It is well known that BS, BH, ATD, and BTD are important processes in the latter channel. BS describes how the low-lying vibrational wave packets move to the region of large nuclear distances in a strong field, leading to the dissociation of the molecule. On the other hand, BH implies that the high-lying vibrational wave packets are localized in the field-induced bound state24. ATD can occur when the nuclear wave packets are excited by absorbing more than one photon. The molecule can also dissociate by absorbing photons with less energy than the dissociation threshold, a process called BTD25. The details of these processes have been widely studied in terms of the nuclear kinetic-energy release (KER) spectrum, a generally accepted method26,27,28,29,30,31,32,33,34. The general numerical method for investigating nuclear dynamics involves solving the TDSE within the BO approximation by considering only the lowest or essential electronic states. However, ultrashort and very intense laser pulses make the BO approximation inadequate for treating the interaction between strong fields and molecules, especially H2+, which contains light nuclei. The dissociation dynamics of H2+ is demonstrably influenced not only by the ionization of H2 but also by the ionization of H2+ itself when initiated from a neutral target35,36,37.
. It is well known that BS, BH, ATD, and BTD are important processes in the latter channel. BS describes how the low-lying vibrational wave packets move to the region of large nuclear distances in a strong field, leading to the dissociation of the molecule. On the other hand, BH implies that the high-lying vibrational wave packets are localized in the field-induced bound state24. ATD can occur when the nuclear wave packets are excited by absorbing more than one photon. The molecule can also dissociate by absorbing photons with less energy than the dissociation threshold, a process called BTD25. The details of these processes have been widely studied in terms of the nuclear kinetic-energy release (KER) spectrum, a generally accepted method26,27,28,29,30,31,32,33,34. The general numerical method for investigating nuclear dynamics involves solving the TDSE within the BO approximation by considering only the lowest or essential electronic states. However, ultrashort and very intense laser pulses make the BO approximation inadequate for treating the interaction between strong fields and molecules, especially H2+, which contains light nuclei. The dissociation dynamics of H2+ is demonstrably influenced not only by the ionization of H2 but also by the ionization of H2+ itself when initiated from a neutral target35,36,37.
Since the landmark experiment of Kling et al. who observed the electron localization on one of the dissociating nuclei of D2+ by utilizing a few-cycle phase-stabilized pulse38, much attention has been paid to electron localization and how energy is shared between the electron and the protons during nuclear dissociation39,40,41,42,43,44,45,46. Recently, Wu and his group studied laser-driven electron-nuclear coupling ingeniously in two-dimensional space, achieving one- or two-dimensionally directional dissociation control and electron localization in diatomic hydrogen system47,48. Most of the directional dissociation controls have been carried out in a 1D space along the direction of laser polarization. In their work, new possibility has been opened to manipulate directional bond breaking of molecules by strong field. Furthermore, they extended experimental measurement to vibrational and orbital resolved electron-nuclear sharing of photon energy for a multielectron CO system even by applying a multicycle laser pulse49. These abovementioned studies confirmed the relative influence of electron and nuclear motions. Except for the experiments on electron localization or asymmetric dissociation, long pulses with a full width at half maximum (FWHM) of tens or hundreds of femtoseconds were applied in most studies50,51,52,53. As a result, the role of nuclear vibrations has been blurred.
The development of laser technology has allowed the achievement of phase-stabilized few-cycle and mid-infrared laser fields. These advanced technologies have been used to study the dynamics of electrons and nuclei. As early as 1991, the pump-probe technique was used to measure the transient ionization spectra of Na2, which probe wave-packet oscillations54. The pump-probe scheme has now become a popular method in laser physics. For example, electron localization after the dissociation of a molecule can be controlled by the pump-probe scheme55,56,57,58. By tuning the time delay of the pump and probe pulses, and analyzing the ionization15,16 and KER28,29,30 signals, the vibrational dynamics of small molecules can be determined.
By numerically solving the TDSE in the presence of nuclear-electron correlation, we studied the dissociation and ionization dynamics of quasi-periodically vibrating H2+ using the pump-probe scheme. The primary aim of this paper is to present an elaborate control of molecular dissociation and ionization, as well as to survey the effect of nuclear motion on ionization and especially on the dissociation of H2+, using advanced few-cycle and mid-infrared laser technologies.
Results and Discussion
Figure 1 outlines the laser fields and the pump-probe scheme used in this work. A two-cycle 2400 nm pump pulse with a relatively weak peak intensity of 1 × 1013 W/cm2 was used to excite vibrations in a nuclear wave packet of H2+ initially prepared at vibrational level υ = 9. Such a weak mid-infrared laser ensures that the molecule do not ionize or dissociate. The chosen initial vibrational state ensured that the internuclear distances R within the quasi-periodically vibrating wave packet did not exceed 7 a.u., hence avoiding CREI. After the pump pulse was applied, the whole nuclear wave packet became a superposition of a few vibrational states close to υ = 9. This result can be verified using a two-state model that includes only the 1sσg and 2pσu states. Most of the nuclear wave packet involves the vibrational components υ = 8, 9, 10, and 11. Next, an intense time-delayed few-cycle laser pulse triggered the nuclear and electronic dynamics as follows. The wavelength, duration, and peak intensity of the delayed laser were 1600 nm, 13.5 fs, and 1 × 1014 W/cm2, respectively59,60. Using this scheme, we could measure the dependences of the dissociation flux, ionization flux, and KER on the time delay, which can be used to probe the dynamics of molecular ionization and dissociation.

(a) Sketch of the pump laser and a time-delayed probe laser. (b) Pump-probe scheme with nuclear wave packets in the dissociation and ionization of H2+.
The time-dependent density of the nuclear wave packet is represented with a color map in Fig. 2a. The time-dependent averages of the internuclear distances and velocities are also shown as white and purple lines, respectively. As shown in Fig. 2b, the dissociation and ionization probabilities modulate with the same period of nuclear vibration. The ionization probability is maximized when internuclear distances reach a maximum value of approximately 5 a.u., where in fact no CREI happens. Interestingly, this trend of ionization probability differs from that reported by Nguyen et al.61, who found maximum intensity in HHG (proportional to the ionization probability) near the time when the internuclear separation is at the equilibrium value, and the nuclei are moving closer. Because the chosen probe-pulse duration was longer in their calculations (FWHM of approximately 14 fs, i.e., the full duration of approximately 38 fs), the nuclear velocity had a strong influence on HHG. Their results imply that ionization occurs during the falling edge of the pulse, when the internuclear distance exceeds the equilibrium value and the nuclei are moving apart. In the case of the few-cycle probe laser used in the present work, ionization almost occurs around the peak of the probe pulse. Therefore, we conclude that the ionization flux in our scheme is related only to the average internuclear distance when ionization occurs, or rather, to the ionization potential, instead of the nuclear velocity.

(a) Time-dependent density of the nuclear wave packet, displayed as a color map. The white and red lines represent the average internuclear distance and velocity, respectively. (b) Dependence of dissociation and ionization probabilities on the time delay. The wavelength, pulse duration, and peak intensity of the delayed probe laser are 1600 nm, 13.5 fs, and 1 × 1014 W/cm2, respectively.
On the other hand, the maximum of the dissociation probability arrives slightly earlier than that of the ionization probability. Although most of the nuclear wavepackets are distributed around the outer turning point when the average internuclear distance reaches the largest value, the dissociation probability is not the maximum. The maximal dissociation probability arrives when the average internuclear distance R is around 4.5 a.u. as marked at time t1 in Fig. 2. At R = 4.5 a.u., the energy difference between the bound and dissociation states is about 2.3 eV which matches the three-photon energy, so the three-photon transition must play an important role and enhance the dissociation62. Meanwhile, one should notice that although the average internuclear distances are both around R = 4.5 a.u. at t1 and t2 in Fig. 2, the dissociation probability at t1 is much larger than that at t2. This means that average velocity will also affect the dissociation process significantly. We attribute the much larger dissociation probability at t1 to the fact that two nuclei are moving away instead of moving close. Thus, we claim for simplicity that the molecules are most likely to dissociate when the two nuclei are moving away at some velocity. The time-delay-dependent dissociation and ionization probabilities with probe intensities of 1 × 1014 and 3 × 1014 W/cm2 are compared in Fig. 3a and b. As the probe-pulse intensity is increased to 3 × 1014 W/cm2, the peak of the dissociation probability is shifted. We attribute this shift to the strong suppression of the dissociation probability of H2+ by ionization50,63. The suppression is much more obvious when the nuclear wavepackets are distributed mostly near the outer turning point. These calculated results demonstrate that the dissociation and ionization dynamics are strongly influenced by the nuclear motion, and therefore the dissociation and ionization of H2+ can be controlled by applying the pump-probe schemes. For example, when the probe pulse is delayed by 108 fs, the ionization probability is almost zero while the dissociation flux remains large at approximately 0.65. It is therefore worthwhile investigating the KER distribution of dissociative nuclei without significant disturbances being introduced by ionization.

(a) Dependence of the dissociation probability on the time delay. (b) Dependence of the ionization probability on the time delay. Note that the ionization probability is multiplied by 20 for the 1 × 1014 W/cm2 peak-intensity data. The other laser parameters are identical to those in Fig. 2.
The remainder of this work focuses on the KER spectra derived from the dissociation channel for a laser intensity of 1 × 1014 W/cm2. The dissociative KER spectra, as functions of the time delay, are plotted in Fig. 4. Three dominant peaks appear periodically in these spectra. The large number of dissociation channels makes it difficult to confirm which channel these peaks in the KER spectra belong to. As mentioned above, after the pump pulse, the nuclear wave packet of H2+ populates the vibrational states υ = 8, 9, 10, and 11. The possible photodissociation pathways via laser-dressed Floquet potentials are presented in Fig. 5. In both diabatic (i.e., with crossing) or adiabatic (with avoided crossing) potential curves, these vibrational levels (υ = 8 ~ 11) are below the threshold for the one-photon channel. Only the υ = 11 state can decay along the one-photon channel by BS. These four states are also above the adiabatic avoided crossing of the three-photon dressed potential. Hence, the nuclear wave packet can dissociate by ATD via (i) the three-photon channel, and (ii) three-photon absorption followed by single-photon release (i.e., a “two-net-photon” channel). The red and green bars on the right of Fig. 4 indicate the regions of the two-net-photon and three-photon dissociation channels, respectively. The time-dependent density of the nuclear wave packets and the KER spectra for the 108 fs delayed probe laser are respectively presented in Fig. 6a and b. The three numbered peaks in Fig. 6b correspond to three jet-like features in Fig. 6a. Peaks 2 and 3 in the KER spectrum arise, respectively, from the two-net-photon and three-photon channels, which have been previously investigated experimentally and theoretically7,64,65.

The red and green bars on the right of the panel indicate the regions of two-net-photon and three-photon dissociation channels, respectively. The laser parameters are the same as in Fig. 2.

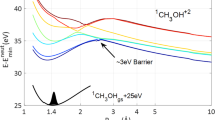
The solid lines represent the adiabatic anticrossing in Floquet theory, whereas the dotted and dot-dashed lines denote the laser-dressed diabatic potential curves. The horizontal lines represent the vibrational levels, υ = 8 ~ 11. The dissociation channels of ATD, BS, and BTD are indicated by the red, green, and blue arrows, respectively.

(a) Time-dependent density of the nuclear wave packet when a 108 fs delayed probe pulse is applied. The white dotted line represents the electric field of the probe laser. The triple jet-like structure corresponds to the three dominant peaks in (b). (b,c) KER spectra produced when the probe pulses are delayed by 108 fs and 115 fs, respectively. (d,e) and (f) describe BTD dynamics. (d) On the leading edge of the probe pulse, a laser-induced adiabatic state is formed and the nuclear wave packet approaches the well. (e) As the intensity increases to the main peak, the gap profile changes and the wave packet is trapped in the 3–0 well. (f) When the intensity of the probe pulse decreases, the curvature of the laser-induced adiabatic potential near R = 6 a.u. changes from upward to downward, so that a part of the trapped wave packet moves to the one-photon dissociation limit. The laser parameters are the same as those of Fig. 2.
The mechanism underlying peak 1 is slightly different from that of the other peaks. The jet-like structure labeled 1 in Fig. 6a appears when the probe pulse is almost ended. The BTD process responsible for this effect is illustrated in Fig. 6d–f. The wave packet is firstly trapped in the potential well formed by the three-photon dressed 2pσu state and the zero-photon undressed 1sσg state (3-0 well). Figure 5 shows the initially populated four vibrational states all being located in this well. At the leading edge of the probe pulse, the nuclear wave packet approaches the laser-induced adiabatic 3-0 well near R = 4.5 a.u. The increased intensity modifies the gap shape near the avoided crossing to a larger internuclear distance. The wave packet is thus trapped in the altered 3-0 well near R = 6.0 a.u., as shown in Fig. 6e. As the laser intensity falls at the trailing edge of the probe pulse, the laser-induced upper adiabatic potential is no longer a well (upward curvature) around R = 6 a.u. but instead curves downward, as shown by the red lines in Fig. 6e and f. A part of the trapped nuclear wave packet then moves to the one-photon dissociation limit. We know from previous studies that peak 3 results from three-photon dissociation, peak 2 from two-net-photon dissociation, and peak 1 from either BTD or two-net-photon dissociation. Furthermore, the dissociation probabilities are almost equal for the cases with delays of 108 or 115 fs (as marked by the horizontal dashed line in Fig. 2b). However, the KER spectra in Fig. 6b and c show significant differences. Because most of the nuclear wave packet is localized at the outer turning point near 4.5 a.u. (Fig. 1a) for the time delay of 115 fs, dissociation from BTD is enhanced; meanwhile, dissociation from ATD via the direct three-photon channel is suppressed, resulting in a higher peak 1 and a lower peak 3.
Conclusion
In summary, we explored the dissociation and ionization dynamics of quasi-periodically vibrating H2+ in an intense few-cycle laser field using quantum-dynamics calculations. Taking advantage of the pump-probe scheme, we obtained the time-delay-dependent dissociation and ionization signals, and also KER spectra. The calculation results demonstrate that the laser-induced dynamics of H2+ is strongly influenced by the nuclear motion and nuclear-electron correlation. It is certain that the ionization flux is only related to the ionization potential but not to the velocity of the nuclei before the internuclear distance reaches a critical value in CREI. The dissociation of H2+ is not maximized when the internuclear distance reaches its maximum, which differs from the case of ionization. The hydrogen molecular ion is most likely to dissociate when the two nuclei are moving away at some velocity. As the intensity of probe laser is increased, competition between dissociation and ionization becomes more apparent. By appropriately choosing the delayed time of the probe pulse, we observed three clear peaks in the KER spectra and explained them in terms of both ATD and BTD. These involved processes being opened and closed periodically, depending on the time delay between the pump and probe pulses. As the nuclear wave packet is mostly localized at the outer turning point, the ATD channel is suppressed while the BTD is enhanced. We expect these findings will prove to be useful in studies of ultrafast dynamics in diatomic molecules, exploiting accessible laser technology. They also provide a useful method for steering the electronic and nuclear dynamics of diatoms by means of intense laser fields.
Methods
All the numerical calculations were carried out using our quantum dynamics program LZH-DICP66. Since the used laser is linearly polarized along with the molecular axis, we reduced the quantum dynamics calculations in 1D nuclear coordinate and 1D electronic coordinate (1D + 1D). Atomic units are used throughout unless otherwise stated, therefore, in the dipole approximation the 1 + 1D NBO TDSE for H2+ can be described as67,68:

where  with a = 0.0 and b = 1.0,
with a = 0.0 and b = 1.0,  ,
,  , E(t) is the electric field, ω is the laser frequency, μR = M/2 and μz = 2 M/(2 M + 1) are the reduced masses with M = 1836.15 a.u., R is internuclear distance, and z is the electron coordinate with respect to the nuclear center of mass. In the simplified coordinate with a softcore Coulomb potential for H2+, the computed dissociation energy (De) of the ground state and ionization potential (Ip) are different from its real values (De = 3.0 eV from present 1D model vs. De = 2.8 eV from full-dimensional calculations or experiments, and at the equilibrium geometry Ip = 31.4 eV from present 1D model vs. Ip = 29.9 eV from full-dimensional calculations or experiments), nevertheless, such differences will not change our main conclusions.
, E(t) is the electric field, ω is the laser frequency, μR = M/2 and μz = 2 M/(2 M + 1) are the reduced masses with M = 1836.15 a.u., R is internuclear distance, and z is the electron coordinate with respect to the nuclear center of mass. In the simplified coordinate with a softcore Coulomb potential for H2+, the computed dissociation energy (De) of the ground state and ionization potential (Ip) are different from its real values (De = 3.0 eV from present 1D model vs. De = 2.8 eV from full-dimensional calculations or experiments, and at the equilibrium geometry Ip = 31.4 eV from present 1D model vs. Ip = 29.9 eV from full-dimensional calculations or experiments), nevertheless, such differences will not change our main conclusions.
We employ the sine basis functions to define a discrete variable representation for the translational coordinates R and z. The time-dependent wave function is advanced using the standard second-order split-operator approach with time steps of 0.2 a.u. and 0.05 a.u. for nuclear and electronic movements respectively. To economize on computation time, we take advantage of the disparity in the time scales of the nuclear and electronic motion. Note that the interaction potential includes all the potential energy of the system plus a purely imaginary term to produce an absorbing boundary. In our calculation, R is extended from 0.1 a.u. to 30 a.u. with a spatial step of ΔR = 0.1 and z grid is from −200 a.u. to 200 a.u. with Δz = 0.2.
The “virtual detector” method66,69 is utilized to detect the dissociation and ionization flux and also to obtain accurate KER spectra. In details, the dissociation flux can be defined as

where Rvd and zvd are the positions of flux analyses for nuclear and electron wave packet. In our calculation, we set Rvd and zvd to be 20 a.u. and 25 a.u., respectively. Similarly, the ionization flux can be defined as

At each time step, the momentum of the dissociation flux passing through Rvd is calculated by

After integration over z and through binning of the momentum values for all time, the momentum distribution for dissociation can be accurately determined. Then, the corresponding KER spectra can be obtained. In addition, the time-dependent nuclear probability density in Fig. 6a is obtained by  . More methodological and computational details are referred to our previous works and the other’s studies on strong fields dynamics and phenomena including KER and HHG66,68,70,71.
. More methodological and computational details are referred to our previous works and the other’s studies on strong fields dynamics and phenomena including KER and HHG66,68,70,71.
Additional Information
How to cite this article: Jiang, S. et al. Dissociation and Ionization of Quasi-Periodically Vibrating H2+ in Intense Few-Cycle Mid-Infrared Laser Fields. Sci. Rep. 7, 42086; doi: 10.1038/srep42086 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Villeneuve, D. M., Ivanov, M. Y. & Corkum, P. B. Enhanced ionization of diatomic molecules in strong laser fields: A classical model. Phys. Rev. A 54, 736 (1996).
Zuo, T. & Bandrauk, A. D. Charge-resonance-enhanced ionization of diatomic molecular ions by intense lasers. Phys. Rev. A 52, R2511 (1995).
Moreno, P., Plaja, L. & Roso, L. Ultrahigh harmonic generation from diatomic molecular ions in highly excited vibrational states. Phys. Rev. A 55, R1593 (1997).
Bandrauk, A. D. & Yu, H. High-order harmonic generation by one-and two-electron molecular ions with intense laser pulses. Phys. Rev. A 59, 539 (1999).
Zavriyev, A. et al. Light-induced vibrational structure in H2 + and D2 + in intense laser fields. Phys. Rev. Lett. 70, 1077 (1993).
Frasinski, L. J. et al. Manipulation of bond hardening in H2 + by chirping of intense femtosecond laser pulses. Phys. Rev. Lett. 83, 3625 (1999).
Jolicard, G. & Atabek, O. Above-threshold-dissociation dynamics of H2 + with short intense laser pulses. Phys. Rev. A 46, 5845 (1992).
Posthumus, J. H. et al. Slow protons as a signature of zero-photon dissociation of H2 + in intense laser fields. J. Phys. B: At. Mol. Opt. Phys. 33, L563 (2000).
Muth-Böhm, J., Becker, A. & Faisal, F. H. M. Suppressed molecular ionization for a class of diatomics in intense femtosecond laser fields. Phys. Rev. Lett. 85, 2280 (2000).
Tong, X. M., Zhao, Z. X. & Lin, C. D. Theory of molecular tunneling ionization. Phys. Rev. A 66, 033402 (2002).
Son S. K. & Chu, S. I. Multielectron effects on the orientation dependence and photoelectron angular distribution of multiphoton ionization of CO2 in strong laser fields. Phys. Rev. A 80, 011403(R) (2009).
Awasthi, M. et al. Single-active-electron approximation for describing molecules in ultrashort laser pulses and its application to molecular hydrogen. Phys. Rev. A 77, 063403 (2008).
Takemoto, N. & Becker, A. Multiple ionization bursts in laser-driven hydrogen molecular ion. Phys. Rev. Lett. 105, 203004 (2010).
Canton, S. E. et al. Direct observation of Young’s double-slit interferences in vibrationally resolved photoionization of diatomic molecules. Proc. Natl. Acad. Sci. USA 108, 7302 (2011).
Yuan, K. J., Bian, X. B. & Bandrauk, A. D. Two-center interference in molecular photoelectron energy spectra with intense attosecond circularly polarized XUV laser pulses. Phys. Rev. A 90, 023407 (2014).
Picón, A. et al. Two-center interferences in photoionization of a dissociating H2 + molecule. Phys. Rev.A 83, 013414 (2011).
Peng, L. Y. et al. Dynamic tunnelling ionization of H2 + in intense fields. J. Phys. B: At. Mol. Opt. Phys. 36, L295 (2003).
Constant, E., Stapelfeldt, H. & Corkum, P. B. Observation of enhanced ionization of molecular ions in intense laser fields. Phys. Rev. Lett. 76, 4140 (1996).
A. Saenz, Enhanced ionization of molecular hydrogen in very strong fields. Phys. Rev. A 61, 051402 (2000).
Bandrauk, A. D. & Ruel, J. Charge-resonance-enhanced ionization of molecular ions in intense laser pulses: Geometric and orientation effects. Phys. Rev. A 59, 2153 (1999).
Xu, H. et al. Experimental observation of the elusive double-peak structure in R-dependent strong-field ionization rate of H2 + . Sci. Rep. 5, 13527 (2015).
Qu, W. X. et al. Nuclear correlation in ionization and harmonic generation of H2 + in short intense laser pulses. Phys. Rev. A 65, 013402 (2001).
Phan, N. L., Truong, T. C. & Nguyen, N. T. Effects of nuclear vibration on the ionization process of H2 + in ultrashort intense laser field. J. Phys.: Conf. Ser. 627, 012014 (2015).
Zavriyev, A. et al. Light-induced vibrational structure in H2 + and D2 + in intense laser fields. Phys. Rev. Lett. 70, 1077 (1993).
Numico, R., Keller, A. & Atabek, O. Nonadiabatic response to short intense laser pulses in dissociation dynamics. Phys. Rev. A 56, 772 (1997).
Trump, C. et al. Pulse-width and isotope effects in femtosecond-pulse strong-field dissociation of H2 + and D2 +, Phys. Rev. A 62, 043411 (2000).
Peng, L. Y. et al. Dissociation of H2 + from a short, intense, infrared laser pulse: proton emission spectra and pulse calibration. J. Phys. B: At. Mol. Opt. Phys. 38, 1727 (2005).
Bocharova, I. A. et al. Time-resolved Coulomb-explosion imaging of nuclear wave-packet dynamics induced in diatomic molecules by intense few-cycle laser pulses. Phys. Rev. A 83, 013417 (2011).
De, S. et al. Tracking nuclear wave-packet dynamics in molecular oxygen ions with few-cycle infrared laser pulses. Phys. Rev. A 82, 013408 (2010).
Ergler, T. et al. Time-resolved imaging and manipulation of H2 fragmentation in intense laser fields. Phys. Rev. Lett. 95, 093001 (2005).
Trump, C., Rottke, H. & Sandner, W. Multiphoton ionization of dissociating D2 + molecules. Phys. Rev. A. 59, 2858 (1999).
Ergler, T. et al. Ultrafast mapping of H2 + (D2 +) nuclear wave packets using time-resolved Coulomb explosion imaging. Phys. B: At. Mol. Opt. Phys. 39, S493 (2006).
Calvert, C. R. et al. Dynamic imaging of a dissociative D2 + nuclear wavepacket in intense laser fields. J. Phys.: Conf. Ser. 58, 379 (2007).
Alnaser, A. S. et al. Simultaneous real-time tracking of wave packets evolving on two different potential curves in H2 + and D2 +, Phys. Rev. A 72, 030702 (2005).
Chelkowski, S., Zuo, T., Atabek, O. & Bandrauk, A. D. Dissociation, ionization, and Coulomb explosion of H2 + in an intense laser field by numerical integration of the time-dependent Schrödinger equation. Phys. Rev. A 52, 2977 (1995).
Walsh, T. D. et al. Laser-induced processes during the Coulomb explosion of H2 in a Ti-sapphire laser pulse. Phys. Rev. A 58, 3922 (1998).
Urbain, X. et al. Intense-laser-field ionization of molecular hydrogen in the tunneling regime and its effect on the vibrational excitation of H2 +. Phys. Rev. Lett. 92, 163004 (2004).
Kling, M. F. et al. Control of electron localization in molecular dissociation. Science 312, 246 (2006).
Tong, X. M. & Lin, C. D. Dynamics of light-field control of molecular dissociation at the few-cycle limit. Phys. Rev. Lett. 98, 123002 (2007).
Kremer, M. et al. Electron localization in molecular fragmentation of H2 by carrier-envelope phase stabilized laser pulses. Phys. Rev. Lett. 103, 213003 (2009).
Wu, J. et al. Electron-nuclear energy sharing in above-threshold multiphoton dissociative ionization of H2 . Phys. Rev. Lett. 111, 023002 (2013).
Madsen, C. B., Anis, F., Madsen, L. B. & Esry, B. D. Multiphoton above threshold effects in strong-field fragmentation. Phys. Rev. Lett. 109, 163003 (2012).
Yue, L. & Madsen, L. B. Inter- and intracycle interference effects in strong-field dissociative ionization. Phys. Rev. A 93, 031401 (2016).
Liu, Y. Q. et al. Selective steering of molecular multiple dissociative channels with strong few-cycle laser pulses. Phys. Rev. Lett. 106, 073004 (2011).
Lan, P. F. et al. Carrier envelope phase dependence of electron localization in the multicycle regime. New J. Phys. 15, 063023 (2013).
Kling, N. G. et al. Carrier-envelope phase control over pathway interference in strong-field dissociation of H2 +. Phys. Rev. Lett. 111, 163004 (2013).
Gong, X. C. et al. Two-dimensional directional proton emission in dissociative ionization of H2 . Phys. Rev. Lett. 113, 203001 (2014).
Lin, K. et al. Directional bond breaking by polarization-gated two-color ultrashort laser pulses. J. Phys. B 49, 025603 (2016).
Zhang, W. et al. Photon energy deposition in strong-field single ionization of multielectron molecules. Phys. Rev. Lett. 117, 103002 (2016).
Ben-Itzhak, I. et al. Dissociation and ionization of H2 + by ultrashort intense laser pulses probed by coincidence 3D momentum imaging. Phys. Rev. Lett. 95, 073002 (2005).
Picón, A., Jaroń-Becker, A. & Becker, A. Enhancement of vibrational excitation and dissociation of H2 + in infrared laser pulses. Phys. Rev. Lett. 109, 163002 (2012).
Sändig, K., Figger, H. & Hänsch, T. W. Dissociation dynamics of H2 + in intense laser fields: investigation of photofragments from single vibrational levels. Phys. Rev. Lett. 85, 4876 (2000).
Pavičić, D., Hänsch, T. W. & Figger, H. Vibrationally resolved strong-field dissociation of D2 + in ion beams. Phys. Rev.A 72, 053413 (2005).
Baumert, T. et al. Femtosecond time-resolved molecular multiphoton ionization: The Na2 system. Phys. Rev. Lett. 67, 3753 (1991).
Yao, H. B. et al. Relative phase control over tunneling ionization of H2 + with a synthesized ω-2ω laser pulse. Phys. Rev. A 90, 063418 (2014).
Wang, Z., Liu, K. L., Lan, P. F. & Lu, P. X. Steering the electron in dissociating H2 + via manipulating two-state population dynamics by a weak low-frequency field. Phys. Rev. A 91, 043419 (2015).
He, F., Ruiz, C. & Becker, A. Control of electron excitation and localization in the dissociation of H2 + and its isotopes using two sequential ultrashort laser pulses. Phys. Rev. Lett. 99, 083002 (2007).
He, F. Control of electron localization in the dissociation of H2 + using orthogonally polarized two-color sequential laser pulses. Phys. Rev. A 86, 063415 (2012).
Guo, J. et al. Influence of vibrational states on high-order-harmonic generation and an isolated attosecond pulse from a N2 molecule. Phys. Rev. A 90, 053410 (2014).
Li, P. C., Liu, I. L. & Chu, S. I. Optimization of three-color laser field for the generation of single ultrashort attosecond pulse. Opt. Express 19, 23857 (2011).
Nguyen, N.-T., Hoang, V.-H. & Le, V.-H. Probing nuclear vibration using high-order harmonic generation. Phys. Rev. A 88, 023824 (2013).
Wu, J. et al. Understanding the role of phase in chemical bond breaking with coincidence angular streaking. Nat. Commun. 4, 2177 (2013).
He, H. X. et al. Dissociation and ionization competing processes for H2 + in intense laser field: Which one is larger? J. Chem. Phys. 136, 024311 (2012).
Orr, P. A. et al. Above threshold dissociation of vibrationally cold HD+ molecules. Phys. Rev. lett. 98, 163001 (2007).
McKenna, J. et al. Controlling strong-field fragmentation of H2 + by temporal effects with few-cycle laser pulses. Phys. Rev. A 85, 023405 (2012).
Lu, R. F., Zhang, P. Y. & Han, K. L. Attosecond-resolution quantum dynamics calculations for atoms and molecules in strong laser fields. Phys. Rev. E 77, 066701 (2008).
Kulander, K. C., Mies, F. H. & Schafer, K. J. Model for studies of laser-induced nonlinear processes in molecules. Phys. Rev. A 53, 2562 (1996).
Lu, R. F. et al. Coherent superposition in the coulomb explosion spectra of H2 +. Chem. Phys. 382, 88 (2011).
Feuerstein, B. & Thumm, U. On the computation of momentum distributions within wavepacket propagation calculations. J. Phys. B: At. Mol. Opt. Phys. 36, 707 (2003).
Yu, C. et al. Intense attosecond pulse generated from a molecular harmonic plateau of H2 + in mid-infrared laser fields. J. Phys. B: At. Mol. Opt. Phys. 47, 055601 (2014).
Feuerstein, B. & Thumm, U. Fragmentation of H2 + in strong 800-nm laser pulses: Initial-vibrational-state dependence. Phys. Rev. A 67, 043405 (2003).
Acknowledgements
This work was supported by NSF of China Grant No. 21373113, the Fundamental Research Funds for the Central Universities of China (Nos 30920140111008 and 30916011105). S.J. gratefully acknowledges the support of Scientific Research Innovation Projects of Jiangsu Province for University Graduate Students with Grant No. KYLX15_0407.
Author information
Authors and Affiliations
Contributions
R.F.L. and C.Y. initiated the project. S.C.J. performed all the calculations with help and discussion with C.Y., G.L.Y. and T.W., S.C.J. and R.F.L. wrote the manuscript. All authors contributed to finalizing and approving the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jiang, S., Yu, C., Yuan, G. et al. Dissociation and Ionization of Quasi-Periodically Vibrating H2+ in Intense Few-Cycle Mid-Infrared Laser Fields. Sci Rep 7, 42086 (2017). https://doi.org/10.1038/srep42086
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42086
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
