
Abstract
Single nucleotide polymorphisms (SNPs) are the most abundant DNA sequence variation in the genomes which can be used to associate genotypic variation to the phenotype. Therefore, availability of a high-density SNP array with uniform genome coverage can advance genetic studies and breeding applications. Here we report the development of a high-density SNP array ‘Axiom_Arachis’ with 58 K SNPs and its utility in groundnut genetic diversity study. In this context, from a total of 163,782 SNPs derived from DNA resequencing and RNA-sequencing of 41 groundnut accessions and wild diploid ancestors, a total of 58,233 unique and informative SNPs were selected for developing the array. In addition to cultivated groundnuts (Arachis hypogaea), fair representation was kept for other diploids (A. duranensis, A. stenosperma, A. cardenasii, A. magna and A. batizocoi). Genotyping of the groundnut ‘Reference Set’ containing 300 genotypes identified 44,424 polymorphic SNPs and genetic diversity analysis provided in-depth insights into the genetic architecture of this material. The availability of the high-density SNP array ‘Axiom_Arachis’ with 58 K SNPs will accelerate the process of high resolution trait genetics and molecular breeding in cultivated groundnut.
Similar content being viewed by others

Introduction
Crop improvement programs in general are focused on enhancing productivity, improving quality and resilience to biotic and abiotic stress by creating and/or harnessing genetic diversity. Genomics-assisted breeding (GAB) has accelerated crop improvement programs for development of improved cultivars in several crops1. Availability of high density genotyping platform with uniformly distributed genome-wide genetic markers is must have genomic resource in a crop for high resolution genetic dissection of complex traits and tracking the favorable alleles in a breeding population2.
Single nucleotide polymorphisms (SNPs) are the most abundant DNA sequence variations among various types of structural/genetic/sequence variations in the genome. Until recently, it has been a tedious, labor-intensive and expensive task to develop even a limited number of SNPs. In the last decade, next-generation sequencing (NGS) technologies have evolved very rapidly and have become the cheapest and fastest method of identification of genome-wide SNPs1. The most commonly used NGS approach for identifying and assaying SNPs is genotyping-by-sequencing (GBS)3. While GBS provides generation of high-density SNP data in less time and less cost, allelic data are not generated for all the SNPs detected among individuals/lines in a given population4. Furthermore, though the imputation methods are available to infer missing data, these methods rely on prior extensive genotyping data.
High-density fixed SNP arrays, though expensive as compared to GBS, provide genotyping data for almost all SNPs for all the individuals in a population. There are several genetics and breeding methods e.g. genomic selection that require consistent genotyping data for the same SNP loci across different germplasm sets and over filial generations in breeding. Therefore, fixed SNP arrays with >40 K SNPs have been developed and used for a variety of genetics and breeding applications in several crops such as rice5,6, maize7,8, sunflower9, soybean10, oat11, cotton12, and wheat13,14.
Groundnut or peanut (Arachis hypogaea) is a globally important crop that is cultivated in >100 countries and consumed in almost every country. The last decade has witnessed a steady increase in demand due to a tremendous population growth in Asia and Africa. In 2014, this crop produced 42.3 Mt from 25.7 Mha with the average productivity of 1649 Kg/ha (http://faostat.fao.org/) but there is an existential need to increase its productivity to meet the growing demand. Cultivated groundnut is an allotetraploid (2n = 4x = 40) species with two subgenomes (A and B) that complicates sharing of diversity between cultivated species and wild diploid species15. To facilitate genetics, breeding as well as evolutionary biology studies, genomic resources such as molecular markers, genetic maps, cytogenetic maps, etc. have been developed in both diploid as well as tetraploid species2,16. Availability of draft genome sequences for both ancestral species of cultivated groundnut namely A. duranensis (A subgenome) and A. ipaensis (B subgenome) in 201617,18 is a major boost for the global groundnut research community.
With the availability of draft genome sequences, re-sequencing and transcriptome sequencing of several accessions of tetraploid species as well as a number of diploid species accessions, we identified a large number of SNPs and selected a comprehensive set of informative genome-wide SNPs. We report here selection and development of array with 58 K SNPs as well as its validation and utility in genetic diversity analysis in groundnut.
Results
SNP selection and array design
The analysis of the sequencing data generated from 41 genotypes (30 tetraploids and 11 diploids) against the genomes of two groundnut progenitors i.e., A. duranensis (A subgenome) and A. ipaensis (B subgenome) (Supplementary Table S1) yielded a total of 163,782 SNPs i.e., 98,375 SNPs from A subgenome and 65,407 SNPs from B subgenome (Fig. 1a). Of the 41 genotypes, sequence analysis of 30 tetraploid genotypes identified 118,860 SNPs (58,438 SNPs from A subgenome and 60,422 SNPs from B subgenome) while 11 diploid genotypes yielded 44,922 SNPs (39,937 SNPs from A subgenome and 4,985 SNPs from B subgenome). Among 30 tetraploid genotypes, analysis of WGRS data for 27 genotypes yielded 113, 835 SNPs (58,438 SNPs from A subgenome and 55,397 SNPs from B subgenome) and RNAseq data of three tetraploid genotypes yielded 5,025 SNPs from B subgenome.

This illustration shows (a) the type of genotypes and sequencing data used for identifying large scale genome-wide SNPs and the final selection of high quality SNPs used in designing SNP array, (b) genomic positions of selected SNPs and (c) annotation of selected 58 K SNPs.
All the identified 163,782 SNPs were subjected to filtering to select SNPs of good quality. The above SNP set also had 52 highly informative SNPs associated with resistance to foliar fungal diseases and oil quality. During filtering process, a total of 96,858 SNPs were discarded as 46,205 SNPs were found on both genomes, 50,642 SNPs were present on either of the two strands of DNA, and 11 SNPs were found identical. As a result, only 66,924 SNPs passed the filtering test (Fig. 1a). From this set, 825 SNPs, however, were further removed because of ambiguity and multi-allelic nature of these SNPs, leaving 66,099 good quality SNPs. From the set of 66,099 good quality SNPs, finally, 58,233 SNPs uniformly distributed across A and B subgenomes were tiled on the Axiom_Arachis array (Supplementary Table S2).
Genomic position and functional annotation of selected SNPs
With respect to genomic positions of the SNPs fixed on Axiom_Arachis array, a total of 22,224 and 23,222 SNPs have come from synonymous coding and intronic regions, respectively (Table 1, Fig. 1b). The other major groups include non-synonymous coding (6,486), UTR_3_prime (4,027) and UTR_5_prime (1,597). In the case of A subgenome, maximum SNPs were located in synonymous_coding (12,087) followed by intronic (11,236), non_synonymous_coding (3,462), UTR_3_prime (2,038) and UTR_5_prime (923) (Table 1). Similarly in the case of B subgenome, maximum SNPs were intronic (11,986) followed by synonymous_coding (10,137), non_synonymous_coding (3,024), UTR_3_prime (1,989) and UTR_5_prime (674) (Table 1).
The functional annotation information was used to categorize the SNPs into different categories i.e., biological processes, molecular function, and cellular component (Fig. 1c). A majority of the SNPs found in genes were classified into cellular component followed by biological process and molecular function. SNPs underlying the genes coding for extracellular, periplasmic space proteins and involved in antioxidant activity were specifically found to be enriched in A. duranensis genome but not in A. ipaensis genome. On the other hand, genes involved in reproductive processes and riboflavin synthase complex were enriched in the A. ipaensis genome but not in the A. duranensis genome. Cell and cell part, binding and catalytic activity, and cellular and metabolic process were the most representative terms in cellular component, molecular function and biological process category.
Genome-wide distribution of selected SNPs
Selected 58,233 SNPs had good representation from both subgenomes of tetraploid groundnut. Of the 58,233 SNPs, 29,983 SNPs have come from A subgenome while 28,250 SNPs from B subgenome and achieved coverage of 2,912 SNPs per pseudomolecule (Table 2). An average 2,998 SNPs per pseudomolecule were selected from A subgenome and the number of SNPs ranged from 2,303 (pseudomolecule A07) to 4,714 (pseudomolecule A01). Similarly B subgenome had an average of 2,825 SNPs per pseudomolecule and ranged from 2,405 (pseudomolecule B01) to 3,443 (pseudomolecule B03). In terms of source for 58,233 SNPs, 44,501 SNPs were selected by comparing both the genome assemblies with tetraploid genotypes, 21 trait linked SNPs for foliar disease resistance and oil quality and 13,732 SNPs with diploid genotypes. Of the 13,732 SNPs, 2,195 SNPs from A. cardenasii, 3,834 SNPs from A. duranensis, 2,389 SNPs from A. stenosperma, 2,605 SNPs from A. magna and 2,709 SNPs from A. batizocoi were identified (Table 3, Supplementary Table S3).
Polymorphism analysis in the ‘Reference Set’
Axiom_Arachis SNP array was used to genotype the ‘Reference Set’ comprising of 300 genotypes developed by International Crops Research Institute for the Semi-Arid Tropics (ICRISAT) (Fig. 2a–e, Supplementary Table S4). Genotyping of this set with the SNP array resulted in generation of genotyping data for 58,233 SNPs on 297 genotypes as QC for allele call failed for three samples. Upon identifying the polymorphic SNPs separately for the diploid species genotypes and tetraploid species genotypes, 40,714 polymorphic SNPs were identified on a panel of 36 wild genotypes while 9,312 polymorphic SNPs in the set of 264 cultivated tetraploid genotypes. Comparison of SNPs identified in the above two sets resulted in the identification of 5,625 common SNPs (Table 2). Subsequently, 44,424 polymorphic SNPs were identified through combined analysis of all the 297 genotypes of the ‘Reference Set’ and were used for further genetic analysis.

This figure shows (a,b) monomorphic SNPs identified in the genotyping data, (c) polymorphic SNPs without heterozygosity i.e., homozygous SNPs, (d) polymorphic SNPs with heterozygosity, (e) genome-wide distribution of SNPs, and (f) pseudomolecule-wise distribution of SNPs on array and polymorphic SNPs in the ‘Reference Set’.
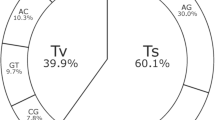
Of the 44,424 polymorphic SNPs, 23,559 SNPs were from A subgenome while 20,865 SNPs from B subgenome and achieved an average of 2,221 polymorphic SNPs per pseudomolecule (Fig. 2f, Table 2). An average 2,356 polymorphic SNPs per pseudomolecule were from A subgenome and the number of SNPs ranged from 1,822 SNPs (pseudomolecule A07) to 3,631 SNPs (pseudomolecule A01). Similarly B subgenome had an average 2,087 polymorphic SNPs per pseudomolecule and it ranged from 1,750 (pseudomolecule B01) to 2,598 (pseudomolecule B03). Further, SNPs selected from A. stenosperma (87.7%) and A. batizocoi (82.1%) showed highest level of polymorphism while SNPs from A. magna (52.5%) showed minimum polymorphism in the ‘Reference Set’ (Table 3). The SNPs from A. hypogaea (76.8%), A. cardenasii (74.9%) and A. duranensis (76.0%) showed similar level of polymorphism (Fig. 3).

This figure shows (a) percentage (%) of SNPs selected from different species using sequencing data for development of array, and (b) percentage (%) of polymorphism achieved by these SNPs in the ‘Reference Set’.
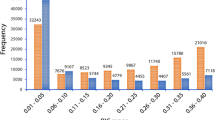
Major allele frequency for the polymorphic SNPs in the ‘Reference Set’ ranged from 0.50 (52 SNPs) to 0.99 (6,853 SNPs) with an average of 0.92 (Supplementary Table S5). Minor allele frequency ranged from zero (1,814 SNPs) to 0.50 (52 SNPs) with an estimated average of 0.08. Similarly, heterozygosity in the population ranged from zero (7,842 SNPs) to 0.87 (AX-147231295) with estimated average of 0.02. Polymorphic information content (PIC) value for SNPs on the array ranged from 0.01 (5,420 SNPs) to 0.50 (608 SNPs) with an estimated average of 0.13 (Supplementary Table S5) in the population.
Genetic analysis of the ‘Reference Set’
Generation of high throughput SNP genotyping data on the ‘Reference Set’ provided an opportunity to gain deeper insights into the genetic relatedness among the genotypes and also the genetic architecture of this important germplasm set (Fig. 4a). The genetic diversity analysis with 44,424 polymorphic SNPs identified four clusters (Cluster-I, Cluster-II, Cluster-III and Cluster-IV) (Fig. 4b, Supplementary Table S6). The Cluster-I consisted of genotypes of the diploid (wild) species while Cluster-II, Cluster-III and Cluster-IV consisted of tetraploid genotypes (A. hypogaea). Among the tetraploid groups, the Cluster-II had genotypes form hypogaea subspecies, Cluster-III had both the subspecies i.e., hypogaea and fastigiata while Cluster-IV had genotypes from fastigiata subspecies. The diverse lines with maximum genetic distance within cultivated groups can be used for developing genetic and breeding populations for both mapping traits as well as for developing improved varieties with desirable agronomic traits and enhanced genetic base.

This figure shows (a) global distribution of genotypes of the ‘Reference Set’. The world map was constructed using cartoDB (https://fee.carto.com/) with OpenStreetMap data (https://www.openstreetmap.org/). (b) Grouping pattern of the ‘Reference Set’ genotypes based on polymorphic SNPs, and (c) genome architecture pattern among different clusters.
This array has also shown significant loss of diversity in the cultivated gene pool and preferential selection of genomic regions in these subspecies (Fig. 4c). The loss of genetic diversity is clearly visible in cultivated genotypes i.e., three clusters (Cluster-II, III and IV) upon comparison with wild species accessions grouped together in Cluster-I. Most importantly, few genomic regions were found conserved and were specific to subspecies fastigiata and hypogaea of the A. hypogaea (cultivated tetraploid). For example, the genomic regions conserved to subspecies fastigiata were observed on pseudomolecules A02, A06, A07 and A10 of A subgenome while B01, B07 and B09 of B subgenome. Similarly, the genomic regions conserved to subspecies hypogaea were observed in pseudomolecules A04 of A subgenome while B04, B08 and B10 of B subgenome.
Identification of subspecies specific high frequency SNPs
We have identified the pseudomolecule-wise distribution of subspecies specific high frequency (>80%) SNPs for subspecies fastigiata and hypogaea (Fig. 5, Supplementary Table S7). Of the total identified 809 high frequency SNPs, 94 SNPs were from subspecies fastigiata and 517 SNPs from subspecies hypogaea. No common SNP was detected between subspecies fastigiata and wild accessions, while, 198 SNPs were found common between subspecies hypogaea and wild accessions. The prediction of effect for these 198 SNPs indicated their location in 133 genes having missense or nonsense mutations. The functions of these genes and their association in various biological functions has been described in the Supplementary Table S8. The enrichment analysis using GO ids showed that majority of genes have binding, catalytic and transporter functions and are involved in catalytic and cellular processes (Supplementary Figure S1).

The frequency of alleles at each SNP locus were calculated and combined chart was prepared using alternate allele frequencies of SNPs with contrasting allele frequencies in three categories, fastigiata, hypogaea and wild. Each bar represents the alternate allele frequency observed at particular SNP site in three categories. The red, blue and green bar/line indicates the alternate allele frequencies observed in fastigiata, hypogaea and wild categories, respectively.
Discussion
High density genotyping ‘Axiom_Arachis’ array
A high density SNP genotyping array with uniform genome coverage is must in any crop for conducting high resolution trait mapping2,19. Development of SNP array for high throughput genotyping was very much required in the case of groundnut due to its large genome size and low genetic diversity in the cultivated gene pool2,16,19. Availability of Axiom_Arachis array with 58,233 SNPs to the Arachis community provides an opportunity to generate high throughput genotyping data on different types of genetic and breeding populations for accelerating genetic diversity, high resolution trait mapping and breeding applications. Similar arrays were developed recently in other crop species such as rice (44 K by McCouch et al.5; 50 K by Chen et al.6; 50 K by Singh et al.20), sunflower (11 K SNPs by Bachlava et al.9), soybean (50 K SNPs by Song et al.10), oil palm (171 K SNPs by Kwong et al.21), maize (58 K SNPs by Ganal et al.7) and wheat (90 K SNPs by Wang et al.13). Much higher density genotyping arrays are available in animal species like chicken (600 K SNPs by Kranis et al.22), cattle (648 K by Rincon et al.23) and human (900 K SNPs by Kathiresan et al.24). In the case of plants, 819 K SNPs arrays for wheat14 and 600 K SNPs arrays for maize8 are the most-dense publicly available genotyping arrays.
Fair representation of genome and Arachis species
The genome size of A and B subgenomes is reported to be 1,070 Mb and 1,360 Mb, respectively17. Axiom_Arachis array developed in this study has fair genome representation i.e., 51.5% from A subgenome and 48.5% from B subgenome with an average 2,998 and 2,825 SNPs per pseudomolecule for A and B subgenome, respectively. Also this array achieved high density genome coverage of 1 SNP per 42 Kb in tetraploid genome while 1 SNP per 36 Kb in A subgenome and 1 SNP per 48 Kb in B subgenome. The above density is comparable to other recently developed SNP arrays in maize7, rice6 and oil palm21.
We also tried to make this array diverse by including informative SNPs from different sources i.e., 76.7% SNPs from A. hypogaea (cultivated tetraploid genotypes), 6.2% SNPs from A. duranensis, 4.7% SNPs from A. batizocoi, 4.5% SNPs from A. magna, 4.1% from A. stenosperma, and 3.8% SNPs from A. cardenasii. The 58,233 SNPs placed on the newly developed array represented mostly intronic region with 39.8% share followed by 38.1% synonymous_coding, 11.1% non-synonymous_coding, 6.9% UTR_3_prime and 2.7% UTR_5_prime. The remaining 1.2% SNPs included intergenic, stop_gained, stop_lost, non-synonymous_start, non-synonymous_stop, start_lost, and others. The 50 K SNP array developed by Singh et al.20 in rice also had large proportion of SNPs from intronic region (41% SNPs) followed by non-synonymous_coding (20%), synonymous_coding (18% SNPs), UTR_3_prime (14% SNPs) and UTR_5_prime (7% SNPs). Functional annotation of SNPs from two subgenomes indicated A. duranensis enriched with genes coding for extracellular, periplasmic space proteins and involved in antioxidant activity while A. ipaensis with genes involved in reproductive processes and riboflavin synthase complex.
Insights on genetic diversity and genetic relationship
The newly developed Axiom_Arachis SNP array was deployed to study genetic diversity and genetic relatedness in the ‘Reference Set’ developed by ICRISAT25. This set has 300 individuals of which 264 are cultivated tetraploid while 36 are wild accessions. Out of 36 wild accessions, 34 were diploid while two accessions were tetraploid belonging to A. monticola. High quality SNP genotyping data was generated successfully for all except three genotypes of the panel mainly due to poor QC for allele call. The polymorphism rate was four times higher in the smaller set of wild genotypes than the larger set of cultivated genotypes with mere ~10% common SNPs between both the sets.
After removing the common SNPs, 77.6% SNPs showed polymorphism in the ‘Reference Set’ with comparatively higher rate of polymorphism in A subgenome (53.0%) than the B subgenome (47%). Considering the genome size of A (1,070 Mb) and B (1,360 Mb) subgenomes, single SNP per 45 Kb polymorphism density has been achieved in A subgenome as compared to single SNP per 65 Kb in B subgenome. Further, SNPs selected from A. stenosperma (87.7%) and A. batizocoi (82.1%) showed highest polymorphism as compared to A. hypogaea (76.8%), A. duranensis (76.0%), A. cardenasii (74.9%) and A. magna (52.5%).
Mean major allele frequency, minor allele frequency, heterozygosity and PIC was found to be 0.92, 0.08, 0.02 and 0.13, respectively in the ‘Reference Set’. Phylogenetic analysis clearly grouped all the genotypes of the ‘Reference Set’ in four groups. As expected, the wild genotypes were grouped together in Cluster-I while cultivated genotypes (A. hypogaea) were clustered into three distinct groups. Majority of the genotypes from hypogaea subspecies were grouped together in Cluster-II, both the subspecies i.e., hypogaea and fastigiata genotypes in Cluster-III and genotypes of fastigiata subspecies in Cluster-IV. The grouping of different subspecies genotypes using the SNP array was found much better than the earlier studies conducted with SSR and DArT genotyping on the same germplasm set26,27.
Conserved genomic regions harboring domestication related genes
The cultivated groundnut crop across the world can be divided into four market types from two subspecies. The subspecies A. hypogaea hypogaea do not flower on main stem, have alternate branching patterns, mature later and produce large seeds while A. hypogaea fastigiata produce flowers on the main stem, have sequential branching patterns, mature earlier and produce smaller seeds28. This array has demonstrated immense power not only in grouping the genotypes of different subspecies of A. hypogaea but also showing preferential selection of genomic regions in these subspecies. Significant loss of diversity can be clearly observed in all three clusters representing cultivated tetraploid genotypes as compared to the cluster representing wild species accessions. This study indicated that during the evolution of subspecies fastigiata and hypogaea of the A. hypogaea (cultivated tetraploid), selective genomic regions remained conserved. These genomic regions might be harboring genes that are responsible for maintaining subspecies specific features.
In order to get further insights, we identified the pseudomolecule-wise subspecies specific high frequency SNPs for fastigiata and hypogaea. Prediction of 198 common SNPs between subspecies hypogaea and wild accessions indicated their location in 133 genes including plant defense against biotic and abiotic stresses, cellular growth and development, seed and pollen development. More importantly genes related to domestication traits such as skotomorphogenesis, flowering time (Aradu.1A8NN.1), seed maturity and germination (Aradu.PZ509.1), lateral root development (Aradu.895HT.1), stem elongation (Aradu.Y6SZD.1) and self-incompatibility (Araip.D2CP3.1) have shown genomic variation between these two subspecies. Such variation for domestication related traits might be playing an important role in retaining the basic features of these two subspecies during the course of evolution.
In summary, the present study reports development of Axiom_Arachis array with 58 K informative SNPs and its successful deployment in understanding the genetic diversity of ICRISAT ‘Reference Set’ in groundnut. This array is an important genomic resource for the Arachis and especially the groundnut community that will be useful not only for accelerating genetics and breeding applications but also to understand evolutionary biology in Arachis species.
Materials and Methods
Plant materials
A total of 41 Arachis spp. accessions (25 from University of Georgia, USA; 15 from ICRISAT, India and one from Crops Research Institute-Guangdong Academy of Agricultural Sciences, Guangzhou, China) were used for generating the sequence data in earlier studies18,29 and the same data was used for identification of SNPs. Of the 41 accessions, 30 genotypes represented cultivated tetraploid species and 11 wild accessions represented 6 different diploid species.
A total of 38 accessions were used for whole genome re-sequencing (WGRS) while 3 accessions (ICGV 91114, JL 24 and J 11, all tetraploids) were used for transcriptome sequencing. 38 accessions used for WGRS included 23 tetraploids, 4 tetraploid pooled samples (resistant and susceptible for foliar disease resistance) and 11 diploid species genotypes representing A. duranensis (PI 475845, ICG 8138 and ICG 8123), A. ipaensis (ICG 8206), A. batizocoi (ICG 8209, ICG 13160 and K9484), A. magna (ICG 8960 and KG30097), A. stenosperma (V10309) and A. cardenasii. The ‘Reference Set’ that is comprised of 300 accessions coming from 48 countries25 including 36 wild species accessions was used for genotyping with SNP array.
DNA/RNA isolation, sequencing and SNP identification
High quality DNA isolation using modified CTAB-based method followed by quantification and quality check of DNA was done as mentioned in Mace et al.30. The WGRS data were generated for 21 tetraploid accessions and 4 diploids at UGA, USA; and 6 tetraploids (TAG 24, GPBD 4 and 4 tetraploid pooled samples for foliar disease resistance) at ICRISAT, India. The RNA-seq data for 3 tetraploid genotypes (ICGV 91114, JL 24 and J 11) were also generated at ICRISAT, India. The WGRS data for 7 diploids (PI 475845, ICG 8138, ICG 8960, ICG 8209, ICG 13160, ICG 8206 and ICG 8123) were generated at Macrogen Inc., South Korea.
The raw sequences obtained were filtered using various softwares to get high quality reads for downstream processing. Briefly, the adapter sequences were trimmed using Cutadapt v1.2.131 while quality trimming was carried out using TrimGalore v0.3.7 software. Such high quality sequences were mapped against the two diploid genomes (A and B subgenomes represented by A. duranensis and A. ipaensis)17 with Bowtie232 and SNPs were identified. Further, the homeologous SNPs were removed using SWEEP Prime version program33. Subsequently, SNPs which were present within 10 kb of A. duranensis and A. ipaensis annotated genes17 were used for downstream processing. The 35 bp sequences flanking to both side of selected SNPs were extracted using custom script and searched against A and B subgenomes for uniqueness using BLASTN program. Finally, the SNPs showing unique hit of ≥94% identity or across at least 60 aligned bases were selected for array development.
Array design using selected SNPs
The selected SNPs representing 10 pseudomolecules each for A subgenome and B subgenome following the above mentioned criteria were subjected to in silico validation. The in silico validation of the assay involved preliminary screening of the designed array file for each selected SNP, including their p-convert values generated using Affymetrix power tool (APT) AxiomGTv1 algorithm to ensure a high-quality final array (http://www.affymetrix.com/estore/partners_programs/programs/developer/tools/powertools.affx). Both forward and reverse probes of each SNP were assigned with p-convert values, derived from a random forest model to predict the probability of SNP conversion on the array. The model considers factors including the probe sequence, binding energy, and expected degree of non-specific hybridization to multiple genomic regions. SNP probes with high p-convert values are expected to convert on the SNP array with a high probability. Potential probes were designed for each SNP in both the forward and reverse direction, each of which was designated as ‘recommended’, ‘neutral’, or ‘not recommended’ based on p-convert values through which the SNP data sets were easily filtered. A SNP marker/strand is recommended if: p-convert >0.6, no wobbles, and poly count = 0. In other words a SNP marker/strand was not recommended if they had duplicate count >0 or poly count >0 or p-convert <0.4 or wobble distance <21, or wobble count > = 3. Therefore, if a marker has the same recommendation for each strand, then it was tiled on strand with the highest p-convert value. None of the [A/T] or [C/G] markers were selected as they take up twice as many features. Finally, probes for selected SNPs were designed and successfully synthesized on the array chip.
Genotyping with the SNP array
Affymetrix GeneTitan®platform was used to genotype “Reference Set” with the SNP array. Initially the target probes were prepared using each DNA sample having minimum quantity of 20 μL of good quality DNA and 10 ng/μL concentration. This procedure is explained in detail in Affymetrix Axiom® 2.0 Assay Manual. These samples were then amplified, fragmented and hybridized on chip followed by single-base extension through DNA ligation and signal amplification. This procedure is explained in detail in Affymetrix Axiom® 2.0 Assay Manual Target Prep Protocol QRC. The GeneTitan® Multi-Channel Instrument was then used for staining and scanning the samples and the details are provided in (http://media.affymetrix.com/support/downloads/manuals/axiom_2_assay_auto_workflow_user_guide.pdf).
SNP allele calling and data analysis
Allele calling was done using Axiom™ Analysis Suite version 1.0 using its three workflows i.e., Best Practices, Sample QC, Genotyping and Summary Only (http://media.affymetrix.com/support/downloads/manuals/axiom_analysis_suite_user_guide.pdf). We used ‘Best Practices’ workflow to perform quality control (QC) analysis of samples to select only those samples which passed the QC test for further analysis. The ‘Sample QC’ workflow was then used to produce genotype calls for the samples which passed QC analysis using ‘Best Practices Workflow’. The ‘Genotyping’ workflow was used to perform genotyping on the imported CEL files regardless of the sample QC matrix. Before making the genotyping calls, samples not passing the QC were removed as their inclusion may reduce the quality of the analyzed results. Finally the ‘Summary Only’ workflow was used to produce a summary containing details on the intensities for the probe sets for use in copy number analysis tools. It also allows to export the SNP data after the analysis is completed for downstream analysis. We have analyzed the diploid and tetraploid genotypes separately keeping the DQC > 0.75 and call rates >90. The above criteria helped in removing the SNPs having low call rates and keeping only the high quality SNPs for the further analysis. NR database was used to annotate all SNP containing genes using BLASTx program (http://blast.ncbi.nlm.nih.gov/blast/Blast.cgi?PROGRAM=blastx&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) with cut off E value <1.0E-5. Further, these SNPs were annotated and their effect on gene function was predicted using SNPEff V4.234 software. For this, a file containing reference genome sequence in FASTA format and general feature format (GFF) file containing co-ordinates of various gene features such as, coding sequence (CDS), 5′ untranslated region (5′ UTR), 3′ untranslated region (3′ UTR), etc. were downloaded from the PeanutBase35 and used to build genome database. The annotation of gene models identified from peanut genome has been described in Bertioli et al.17.
Diversity analysis
Called allelic data was used for studying genetic diversity and genetic relationship among individuals of the “Reference Set”. The polymorphic information content (PIC), major allele frequency, number of observations, availability and gene diversity were calculated using the software PowerMarker ver. 3.2536.
Identification of subspecies specific high frequency SNPs
In order to identify subspecies specific SNPs, the genotypes from the ‘Reference Set’ were divided into three categories viz. fastigiata, hypogaea and wild accessions. The allele frequency at each SNP site within each category was calculated. Further, the allele frequencies at each SNP position were compared between three categories and SNPs with contrasting allele frequencies in all the three categories were identified. Additionally, the annotations of these SNPs and their effect on genes were predicted using SNPEff V4.234 and SNPs with moderate (missense mutations) and high (nonsense mutations) effect were identified. Further, the protein coding sequences of genes containing missense and nonsense SNPs were extracted and the functional annotation and gene ontology analysis was carried out using BlastGO37 software to determine their involvement in particular biological function.
Additional Information
How to cite this article: Pandey, M. K. et al. Development and Evaluation of a High Density Genotyping ‘Axiom_Arachis’ Array with 58 K SNPs for Accelerating Genetics and Breeding in Groundnut. Sci. Rep. 7, 40577; doi: 10.1038/srep40577 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Varshney, R. K. et al. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 27, 522–530 (2009).
Varshney, R. K. et al. Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol. Adv. 31, 1120–1\134 (2013).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, e19379 (2011).
Jaganathan, D. et al. Genotyping-by-sequencing based intra-specific genetic map refines a “QTL-hotspot” region for drought tolerance in chickpea. Mol. Genet. Genomics 290, 559–571 (2015).
McCouch, S. R. et al. Development of genome-wide SNP assays for rice. Breed. Sci. 60, 524–535 (2010).
Chen, H. et al. A high density SNP genotyping array for rice biology and molecular breeding. Mol. Plant. 7, 541–553 (2014).
Ganal, M. W. et al. A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS One 6, e28334 (2011).
Unterseer, S. et al. A powerful tool for genome analysis in maize: development and evaluation of the high density 600 k SNP genotyping array. BMC Genomics 15, 823 (2014).
Bachlava, E. et al. SNP discovery and development of a high-density genotyping array for sunflower. PLoS One 7, e29814 (2012).
Song, Q. et al. Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS One 8, e54985 (2013).
Tinker, N. A. et al. A SNP genotyping array for hexaploid oat. The Plant Genome 7, doi: 10.3835/plantgenome2014.03.0010 (2014).
Hulse-Kemp A. et al. Development of a 63K SNP array for cotton and high-density mapping of intraspecific and interspecific populations of Gossypium spp. G3 (Bethesda) 5, 1187–1209 (2015).
Wang, S. et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 12, 787–796 (2014).
Winfield, M. O. et al. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 14, 1195–1206 (2016).
Simpson, C. E., Krapovickas, A. & Valls, J. F. M. History of Arachis including evidence of A. hypogaea L. progenitors. Peanut Sci. 28, 78–80 (2001).
Pandey, M. K. et al. Advances in Arachis genomics for peanut improvement. Biotechnol. Adv. 30, 639–651 (2012).
Bertioli, D. J. et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 48, 438–446 (2016).
Chen, X. et al. Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis and allergens. Proc. Natl. Acad. Sci. USA 113, 6785–6790 (2016).
Pandey, M. K. et al. Emerging genomic tools for legume breeding: current status and future prospects. Front. Plant Sci. 7, 455 (2016).
Singh, N. et al. Single-copy gene based 50 K SNP chip for genetic studies and molecular breeding in rice. Sci. Reports 5, 11600 (2015).
Kwong, Q. B. et al. Development and validation of a high density SNP genotyping array for African oil palm. Mol. Plant. 9, 1132–1141 (2016).
Kranis, A. et al. Development of a high density 600 K SNP genotyping array for chicken. BMC Genomics 14, 59 (2013).
Rincon, G. et al. Performance of bovine high-density genotyping platforms in Holsteins and Jerseys. J. Dairy Sci. 94, 6116–6121 (2011).
Kathiresan, S. et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 41, 334–341 (2009).
Upadhyaya, H. D., Ortiz, R., Brame, P. J. & Singh, S. Development of a groundnut core collection using taxonomical, geographical, and morphological descriptors. Genet. Resour. Crop Evol. 50, 139–148 (2003).
Khera, P. et al. Single nucleotide polymorphism–based genetic diversity in the reference set of peanut (Arachis spp.) by developing and applying cost-effective kompetitive allele specific polymerase chain reaction genotyping assays. The Plant Genome 6, 3 (2013).
Pandey, M. K. et al. Genome-wide association studies for 50 agronomic traits in peanut using the ‘reference set’ comprising 300 genotypes from 48 countries of semi-arid tropics of the world. PLoS One 9, e113326 (2014).
Knauft, D. A. & Gorbet, D. W. Genetic diversity among peanut cultivars. Crop Sci. 29, 1417–1422 (1989).
Holbrook, C. et al. Development and phenotyping of recombinant inbred line (RIL) populations for peanut (Arachis hypogaea). Peanut Sci. 40, 89–94 (2013).
Mace, E. S. et al. A high-throughput DNA extraction protocol for tropical molecular breeding programs. Plant Mol. Biol. Rep. 21, 459–460 (2003).
Marcel, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J, 17(1), 10–12 (2011).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Clevenger, J. & Ozias-Akins, P. SWEEP: A tool for filtering high-quality SNPs in polyploid crops. G3 (Bethesda) 5, 1797–1803 (2015).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Dash, S. et al. PeanutBase and other bioinformatic resources for peanut. (ed. Stalker, H. T. & Wilson, R. F. ) 241–252 (AOCS Press, 2016).
Liu, K. & Muse, S. V. PowerMarker: integrate analysis environment for genetic marker data. Bioinformatics 21, 2128–2129 (2005).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Acknowledgements
The authors are thankful to Bill & Melinda Gates Foundation (Tropical Legumes III), MARS Chocolate Inc., and The Peanut Foundation for financial assistance. The work reported in this article was undertaken as a part of the CGIAR Research Program on Grain Legumes. ICRISAT is a member of the CGIAR.
Author information
Authors and Affiliations
Contributions
M.K.P. performed most of the experiments; G.A., J.C. and C.C. selected and filtered SNPs for designing array; M.K.P., S.M.K., S.N.N., M.S. and M.K.V. analyzed genotyping data and conducted genetic analysis; A.C., X.L., X.C., B.G., S.L.-B., D.J.B., S.A.J., P.O.-A., and H.D.U. contributed genetic material and sequencing data; M.K.P. and R.K.V. interpreted the results and wrote the manuscript; R.K.V. conceived, designed and supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pandey, M., Agarwal, G., Kale, S. et al. Development and Evaluation of a High Density Genotyping ‘Axiom_Arachis’ Array with 58 K SNPs for Accelerating Genetics and Breeding in Groundnut. Sci Rep 7, 40577 (2017). https://doi.org/10.1038/srep40577
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40577
This article is cited by
-
High-throughput diagnostic markers for foliar fungal disease resistance and high oleic acid content in groundnut
BMC Plant Biology (2024)
-
Genetic diversity assessment of groundnut (Arachis hypogaea L.) for polyphenol content and antioxidant activity: unlocking the nutritional potential
Journal of Plant Biochemistry and Biotechnology (2024)
-
Novel Microsatellite Markers Derived from Arachis pintoi Transcriptome Sequencing for Cross-Species Transferability and Varietal Identification
Plant Molecular Biology Reporter (2024)
-
Designing future peanut: the power of genomics-assisted breeding
Theoretical and Applied Genetics (2024)
-
Genome-wide association studies reveal novel loci for resistance to groundnut rosette disease in the African core groundnut collection
Theoretical and Applied Genetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
