
Abstract
Genotyping-by-Sequencing (GBS) was applied in a set of 53 diploid Prunus rootstocks and five scion cultivars from three subgenera (Amygdalus, Prunus and Cerasus) for genome-wide SNP identification and to assess genetic diversity of both Chilean and Spanish germplasm collections. A group of 45,382 high quality SNPs (MAF >0.05; missing data <5%) were selected for analysis of this group of 58 accessions. These SNPs were distributed in genic and intergenic regions in the eight pseudomolecules of the peach genome (Peach v2.0), with an average of 53% located in exonic regions. The genetic diversity detected among the studied accessions divided them in three groups, which are in agreement with their current taxonomic classification. SNPs were classified based on their putative effect on annotated genes and KOG analysis was carried out to provide a deeper understanding of the function of 119 genes affected by high-impact SNPs. Results demonstrate the high utility for Prunus rootstocks identification and studies of diversity in Prunus species. Also, given the high number of SNPs identified in exonic regions, this strategy represents an important tool for finding candidate genes underlying traits of interest and potential functional markers for use in marker-assisted selection.
Similar content being viewed by others

Introduction
Prunus is a genus belonging to the subfamily Prunoideae of the family Rosaceae1. Several species of this large genus, known as stone fruits, are among the most important for the world fruit industry, providing edible and tasty fruits highly appreciated by consumers (e.g., peaches, plums, cherries, apricots and almonds). Cherries and plums are well adapted to the cooler temperate areas of the world, while almonds, apricots and peaches are grown in warmer temperate regions with Mediterranean climate. Nevertheless, all of them require adequate winter chilling to achieve an effective fruit set and production2.
In modern stone fruit production, trees require to be grafted onto a rootstock well adapted to the soil most prevalent conditions. It means that the tree is composed of two genetically distinct partners joined by grafting of the aerial part (scion) on the rootstock, the later including part of the trunk and roots. The scion is the fruiting cultivar while the rootstock is responsible for water and nutrient uptake. In addition, some of the most important agricultural traits of the tree may be substantially influenced by the rootstock3,4,5,6, such as blossom, fruit set, size, sugars and other fruit quality parameters, as well as tolerance to biotic and abiotic stresses. While new stone fruit scion cultivars development is addressed by numerous breeding programs around the world, only very few of them aim to the development of new Prunus rootstocks.
In Chile, the ‘Centro de Estudios Avanzados en Fruticultura’ (CEAF) started a Prunus rootstock breeding program in 2010, with collaboration of the ‘Estación Experimental de Aula Dei-Consejo Superior de Investigaciones Científicas’ (EEAD-CSIC). From 1950, the survey and establishment of Spanish Prunus germplasm collections were conducted by EEAD-CSIC aiming to preserve and use this material in breeding programs to obtain new stone fruit rootstocks, with specific adaptation to Mediterranean environments7. Furthermore, new rootstocks generated by CEAF and EEAD-CSIC have the potential to be graft-compatible with scions from different species8, which is a desirable characteristic for stone fruit producers. Therefore, better understanding of the molecular background of the material currently released by and/or used in rootstock breeding programs is of great importance.
Effective utilization of Prunus rootstocks in breeding programs depends upon accurate and unambiguous characterization9. In addition, the knowledge of the genetic diversity and relationships among the cultivated species of Prunus is important to recognize gene pools, to identify pitfalls in germplasm collections, and to develop effective conservation and management strategies2. Current trends in breeding stone fruit rootstocks are based on the production of interspecific hybrids, aiming at combining favorable traits from different species7,10. Genetic characterization of diversity and relationships at the interspecific level include studies on the systematic relationships within Prunus using allozyme polymorphisms11, chloroplast DNA variation12,13,14, Internal Transcribed Spacer (ITS) sequence variation of nuclear ribosomal DNA15, ITS and chloroplast trnL–trnF spacer sequence variation16 and Amplified Fragment Length Polymorphisms (AFLPs)2. Molecular characterizations and estimation of relationship specifically between Prunus rootstocks have been performed using molecular markers, such as Random Amplification of Polymorphic DNA (RAPD)17,18 and Single Sequence Repeats (SSRs)9,19,20,21. In recent years, advances in next-generation sequencing (NGS) have enabled the use of Single-Nucleotide Polymorphisms (SNPs) as other important type of molecular marker.
In the last decade, SNPs have become the markers of choice in molecular genetics due to their frequency in genomes and high-throughput, cost effectiveness for their detection using various approaches and platforms22,23. SNPs have been indicated as the major factors in the creation of phenotypic variation and their effect on functional changes of genes is used as a tool in functional genomics of organisms24. For Prunus species, the availability of the peach genome sequence25,26 enables anchoring of SNPs identified through NGS to corresponding positions in the genome, identification of SNP-carrying genes and prediction of the effect of SNPs.
Genotyping-by-Sequencing (GBS) is one of the NGS approaches, which enables the simultaneous discovery and genotyping of thousands of SNPs in a set of multiplexed samples27. In this approach, single and/or double-digestion of DNA with restriction enzymes is used to produce a reduced representation of the genome of each sample27,28. Single and/or double-digest GBS generate massive datasets of SNPs for a range of applications and is widely applicable in both model and non-model organisms29,30,31. It has been described that, compared with single-digest GBS, double-digest protocol greatly simplifies quantification of the library prior to sequencing and could generate a suitable and uniform complexity reduction of the genome28.
In Prunus species, single-digest GBS has been used for identification of a high number of SNPs for linkage maps construction32,33,34,35 and analysis of population genetic structure36,37,38. The use of double-digest GBS in Prunus has not been reported as far as the authors know. Also, and despite their importance, molecular characterization of Prunus rootstocks using GBS or other NGS tools has not been published. In this work, we used double-digest GBS for the identification of SNPs from 58 diploid accessions, most of them rootstocks and interspecific hybrids, members of three different Prunus subgenera (Amygdalus, Prunus and Cerasus, plus Prunus-Amygdalus hybrids). Some of these accessions are representatives of the most important breeding programs of the world (released during the last two decades), such as INRA (France) and UC Davis (USA) programs, and they are extensively used by the fruit industry. Genetic relationship between accessions, the prediction of SNP effects and the identification of SNP-carrying genes is presented. The usefulness of the presented information in understanding the genomic and phenotypic differences among Prunus accessions and its potential towards substantial improvement in knowledge about the genome structure of accessions from Prunus subgenera is discussed.
Results
High-throughput genotyping of Prunus accessions
Double-digest GBS produced between 6,801,412 and 14,293,180 read pairs, with an average of 11,117,746 reads per individual. This extremely deep sequencing led to a mean depth per SNP of 1,323 across the entire dataset (Supplementary Table S1). The number of unique tags varied among 2,174,744 and 4,952,765, with an average of 3,651,464 tags per individual. A total of 45,382 high quality SNPs (MAF > 0.05; missing data < 5%), evenly distributed over the eight pseudomolecules of peach (Pp01 to Pp08), were identified. The number of identified SNPs ranged from 4,122 for Pp08 to 10,762 for Pp01 (Table 1). A total of 224.5 Mb (99.4%) of the peach genome was covered with marker density of approximately one SNP per 5 Kb. Gaps were observed in all pseudomolecules, with the largest gap per pseudomolecule ranging from 456 Kb (Pp06) to 1.3 Mb (Pp04). Physical position of each SNP along peach pseudomolecules allowed the identification of common markers with the cherry 6 K SNP array v139 and IRSC 9 K peach SNP array v140 that were updated with the Peach v2.0 as a reference genome (www.rosaceae.org)41. Only a group of 49 SNPs were in common between our study and the cherry 6 K SNP array v1, while 75 SNPs were common between our study and the IRSC 9 K peach SNP array v1 (data not shown).
Based on the nucleotide substitution, SNPs were classified into transitions (Ts) and transversions (Tv) (Fig. 1). Transitions were observed in 27,236 (60.1%) and transversions in 18,146 SNPs (39.9%). The frequency of substitutions was 13,639 (30.1%) C/T, 13,597 (30.0%) A/G, 5,497 (12.1%) A/T, 4,697 (10.3%) A/C, 4,394 (9.7%) G/T, and 3,558 (7.8%) C/G, with the transitions to transversions ratio of 1.5.

Classification of SNPs based on their nucleotide substitutions, either transitions (Ts) or transversions (Tv).
Percentage of heterozygous positions is presented in Fig. 2 and Supplementary Table S2. Accessions showing the lowest percentage of heterozygous sites were ‘Pomona’ (0.35%), ‘Nemaguard’ (0.51%) and ‘Nemared’ (0.61%), all members of the Amygdalus subgenus, while those showing the highest percentage of heterozygous sites were the Prunus-Amygdalus hybrids AG-‘030104’ (41.22%), ‘AG-030107’ (41.23%) and ‘R R’ (41.64%). The two duplicates of ‘Adara’ and ‘Citation’ shared similar results, while the two ‘Mariana 2624’ samples were not concordant.

Percentage of heterozygous positions for 58 Prunus accessions used in the study. Mar 2624 CEAF - ‘Mariana 2624’ CEAF; Mar 2624 CSIC - ‘Mariana 2624’ CSIC; Myrob 713AD – ‘Myrobalan 713AD’; Myrob 2201 – ‘Myrobalan 2201’; Myrob 2261 – ‘Myrobalan 2261’; Myrob B – ‘Myrobalan B’; Myrob GF3-1 – ‘Myrobalan GF3-1’; Myrob m2 – ‘Myrobalan m2’; N cherry – Nanking cherry; Nemag m1 – ‘Nemaguard’ m1; R 20 – ‘Densipac’; R 40 – ‘Nanopac’; R 70 – ‘Purplepac’; R 90 – ‘Greenpac’; R R – ‘Replantpac’; T de Litera – ‘Tamarite de Litera’; T × N – ‘Titan × Nemared’.
Phylogenetic analysis
An UPGMA dendrogram grouped accessions into three major clusters, which, in most cases, were in agreement with their taxonomic classification (subgenera Amygdalus, Prunus and Cerasus; Table 2 and Fig. 3). Cluster Amygdalus consisted of 31 accessions divided in two subclusters, one formed by four accessions (Subcluster A) and other formed by 27 accessions (Subcluster B). Subcluster A was comprised of four accessions, all Prunus-Amygdalus hybrids. Subcluster B is divided in two groups, B1, with 25 accessions, and B2, with two accessions. Group B1 is further divided in two subgroups, with ‘Carmel’ (P. dulcis), and ‘Hansen 2168’ and ‘Hansen 536’ (both P. dulcis × P. persica hybrids) being separated from the main subgroup. All accessions in the main subgroup have P. persica in their genetic background. Accessions with P. davidiana in their genetic background were grouped more tightly, separated from accessions with P. dulcis in their background. Group B2 is formed by two samples of ‘Citation’, a Prunus-Amygdalus hybrid (P. salicina × P. persica), one from CEAF and the other one from CSIC.

Phylogenetic analysis of 58 Prunus accessions generated through the UPGMA method. Mar 2624 CEAF - ‘Mariana 2624’ CEAF; Mar 2624 CSIC - ‘Mariana 2624’ CSIC; Myrob 713AD – ‘Myrobalan 713AD’; Myrob 2201 – ‘Myrobalan 2201’; Myrob 2261 – ‘Myrobalan 2261’; Myrob B – ‘Myrobalan B’; Myrob GF3-1 – ‘Myrobalan GF3-1’; Myrob m2 – ‘Myrobalan m2’; N cherry – Nanking cherry; Nemag m1 – ‘Nemaguard’ m1; R 20 – ‘Densipac’; R 40 – ‘Nanopac’; R 70 – ‘Purplepac’; R 90 – ‘Greenpac’; R R – ‘Replantpac’; T de Litera – ‘Tamarite de Litera’; T × N – ‘Titan × Nemared’.
Cluster Prunus is comprised of 22 accessions, divided in Subcluster C, with three accessions, and Subcluster D, with 19 accessions. Subcluster C is formed by Prunus-Amygdalus hybrids [P. cerasifera × (P. persica × P. dulcis)]. In the Subcluster D, accessions with P. cerasifera in their genetic background are grouped separately from other accessions. Two unexpected results were observed in the Subcluster D, with ‘Larry Ann’ not grouping with ‘Angeleno’, both P. salicina, but instead grouping with ‘Mariana 2624’ (P. cerasifera × P. munsoniana) accessions. Also, ‘Mariana 2624’ accessions from both CEAF and CSIC were clustered together, but results suggest that they are not the same accessions. Two interspecific hybrids, ‘R 20’ (P. besseyi × P. cerasifera) and ‘Isthara’ [(P. cerasifera × P. salicina) × (P. cerasifera × P. persica)], were grouped in this subcluster. Nanking cherry (P. tomentosa) was also grouped in the Subcluster D, although it was expected to be grouped with accessions from subgenus Cerasus.
Cluster Cerasus was comprised of five accessions, with ‘Bing’ and ‘Mazzard F12/1’ (P. avium) being closely clustered and separated from ‘Pontaleb’ (P. mahaleb). ‘Maxma 14’ and ‘Maxma 60’, interspecific hybrids considered to be of P. mahaleb × P. avium parentage, were grouped with P. avium accessions.
Population structure analysis
Population genetic structure among the studied Prunus accessions suggested the maximum ∆K-value of K = 3 (Fig. 4), as seen in phylogenetic analysis (Fig. 3). The structure analysis grouped 25 accessions in one population (Amygdalus), 17 in the second population (Prunus) and five in the third population (Cerasus). Eleven accessions were classified as admixed and nine of these accessions showed approximately 50% of membership to both Prunus and Amygdalus populations (Supplementary Table S3). As expected, ‘Ishtara’ showed 76% of membership to the Prunus population and 24% to the Amygdalus population. An interesting result was observed for Nanking cherry, which showed 45% of membership to Prunus, 36% to Amygdalus and 19% to Cerasus. The two duplicates samples of ‘Mariana 2624’ showed a different percentage of membership (90% for ‘Mariana 2624’ CEAF and 93% for ‘Mariana 2624’ CSIC), while the duplicates of ‘Adara’ and ‘Citation’ were identical.

Estimation of the population structure for 58 Prunus accessions from subgenera Amygdalus, Cerasus and Prunus, as well as hybrids between these subgenera. Vertical bars along the horizontal axis represent accessions classified in their estimated membership in each population with the optimal population number K = 3. Three different colors represent different populations, which are related with three Prunus subgenera, Amygdalus (blue), Prunus (green) and Cerasus (red). N cherry – Nanking cherry; R 20 – ‘Densipac’; R 40 – ‘Nanopac’; R 70 – ‘Purplepac’; R 90 – ‘Greenpac’; R R – ‘Replantpac’; T de Litera – ‘Tamarite de Litera’; T × N – ‘Titan × Nemared’. A group of accessions with colored segments, indicates their admixed origin.
Principal components analysis
The first two components of a principal component analysis (PCA) described 55.2% and 21.4% of the variance, respectively. Results supported phylogenetic (Fig. 3) and population structure analyses (Fig. 4). A cluster of Prunus-Amygdalus accessions was located in the central area of the PC1 and PC2 plot, and the Amygdalus, Prunus and Cerasus clusters diverged from it (Fig. 5). Three accessions are individually positioned and separated from these groups: ‘Pontaleb’ (P. mahaleb), in the vicinity of cluster Cerasus; ‘Ishtara’, near members of subgenus Prunus; and Nanking cherry, in the middle, closer to the cluster Prunus than to the cluster Cerasus.

Principal components analysis (PCA) plot using 45,382 SNPs for 58 Prunus accessions. First and second principal components are shown and the proportion of the variance explained by each principal component is indicated in parenthesis. Colors refer to K = 3 genetic populations following Structure analyses; Amygdalus (blue); Prunus (green); Cerasus (red); admixed accessions (gray). N cherry – Nanking cherry.
Classification of SNPs based on the positions on the peach genome
Detailed analysis of the classification of SNPs along the eight peach pseudomolecules was performed using data from only 55 unique accessions; one of the two samples of the accessions analyzed in duplicate was included in the study; e.g., ‘Adara’ CSIC, ‘Citation’ CEAF and ‘Mariana 2624’ CSIC.
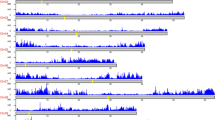
SNP location and gene density (gray) along the eight pseudomolecules of the peach genome, visualized in Circos plot, are presented in Fig. 6. SNP density was determined for the whole group of 55 accessions (purple) and for each subgenus (Amygdalus in blue, Cerasus in red and Prunus in green). Prunus-Amygdalus hybrids and Nanking cherry were included only in the analysis of 55 accessions. A non-uniform pattern of SNP distribution along the eight pseudomolecules was observed when all 55 accessions were considered as well as when accessions within a subgenus were considered. SNP distribution along pseudomolecules was correlated with gene distribution.

SNP distribution and gene density on the Peach v2.0 pseudomolecules. Gene and SNP density were plotted in 1 Mbp sliding window using Circos. Tracks from outside to inside are: distribution of genes on peach genome sequence; pseudomolecules of peach; and four histogram circles of SNPs distribution for 55 Prunus accessions (purple), 25 accessions from Amygdalus subgenus (blue), 5 from Cerasus subgenus (red) and 15 from Prunus subgenus (green).
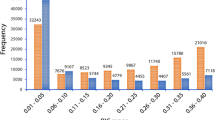
From the initial set of 45,382 SNPs identified, 4,302 (9.7%) were detected in intergenic regions (Fig. 7 and Supplementary Table S4). Proportion of SNPs located in the intergenic regions ranged from 6.5% in Pp05 to 25.3% in Pp04, with 1,647 SNPs (corresponding to 38.3% of SNPs in intergenic regions) located in putative promoter regions (considering 1,000 bp upstream of the transcription start site; Supplementary Table S5). The majority of SNPs (41,080 SNPs, 90.3%) were located in genic regions (53.0% in exons, 27.3% in introns, 4.0% in 5′UTR and 6.0% in 3′UTR). Distribution of SNPs located in exons varied between 43.8–56.4% in Pp04 and Pp08, respectively; in introns between 24.0% for Pp04 and 29.5% for Pp01; in 5′UTR regions between 2.5% for Pp04 and 4.7% for Pp03; and in 3′UTR region between 4.4% for Pp04 and 8.2% for Pp02 (Fig. 7 and Supplementary Table S4). The lowest number of SNPs in genic regions (74.7%) was observed on Pp04, while the highest (93.5%) was observed on Pp05. The 41,080 of SNPs located in genic sequences were present in 4,884 different genes (or 18.2% of genes identified in the peach genome sequence), with an average of 8.4 variants per gene. No significant differences were found when the percentage of SNPs located in genic and intergenic regions for each subgenus were compared to each other and with those identified for the group of 55 accessions (Supplementary Table S6).

Distribution of SNPs in genic [exonic, intronic, 5′- and 3′- untranslated (UTR)] and intergenic regions using the physical position of each SNP on Peach v2.026. Figure represents coverage of each SNP category per pseudomolecule and an average of all categories across all pseudomolecules.
SNP effect prediction
Classification of 45,382 SNPs based on their putative effect on annotated genes, predicted most of the SNPs with a modifier effect (76.0% of the SNPs with impact on noncoding regions); followed by SNPs with a low (14% of the SNPs with synonymous substitution); moderate (9.9% of the SNPs could have a non-synonymous substitution); and high impact (0.1% of the SNPs with disruptive impact on the protein). SNPs with a modifier effect had a downstream gene variant, with a default length of 5 kbp downstream of the most distal polyA addition site, as the most frequent variant (39.0%) (Supplementary Table S7). The most frequent mutations for SNPs with a low effect were synonymous variants (91.4%), while missense variants (99.1%) were the most frequent mutation within SNPs with a moderate effect. Finally, SNPs with a high impact had stop gained variants as the most frequent mutation (57.8%).
SNPs classified as having moderate and high impact were further analyzed in more detail. The missense variant of the SNPs with moderate effect were classified according to the predicted changes in the charges or polarity of the amino acid residues they could cause (Fig. 8). Nonpolar to nonpolar substitutions, which do not alter the properties of amino acid residues, were the most frequent variant. Polar to nonpolar and nonpolar to polar substitutions, which cause changes in polarity, were the second and third more frequent variants, respectively. Less frequent substitutions were basic to acid variations, which are related with changes in charges of amino acid residues.

Substitution patterns of the properties of amino acid residues caused by SNPs.
A group of 128 SNPs with potential to generate a high impact on gene function and phenotype of the plant material analyzed in this study were identified (Supplementary Tables S8 and S9). This group of SNPs affected 119 genes in the peach genome, with at least one SNP in most of the genes, except Prupe.1G243700, Prupe.1G433200, Prupe.2G030900, Prupe.2G230500, Prupe.5G138700, Prupe.6G273700 and Prupe.8G211800, which had two, and Prupe.5G026300, with three SNPs (Supplementary Table S8). A group of 94 SNPs was present in coding DNA sequences (exons), 29 in introns, two in 5′UTR and three in 3′UTR. The most frequent predicted effect caused by SNPs with a high impact was stop gained (72 SNPs), followed by splice donor and acceptor variants (43 SNPs in total) and stop lost (12 SNPs). Details about genes affected by high impact SNPs are presented in Supplementary Table S8. The variations observed in 55 accessions caused by 128 high impact SNPs are presented in Supplementary Table S9, including the pseudomolecule where each SNP is located, the physical position on the pseudomolecule, the gene affected by the SNP, location of the SNP (exon, intron, etc.) and the change observed using the peach genome as reference. For most of these SNPs, accessions from the same subgenus exhibited the same genotype which is either matching the reference or not. For example, in Prupe.1G152100 Amygdalus accessions had the same SNP as the reference (C/C), while Prunus and Cerasus accessions show the variation (T/T). Out of the 119 genes, only 36% had hits in KOG database (Fig. 9). Most of the genes were associated with metabolism (35%), with lipid transport and metabolism as the most frequent function, followed by genes related with cellular processes and signaling (28%), and information storage and processing (15%), while 23% of these genes were poorly characterized.

KOG-annotation-based classification of genes harboring high impact SNPs.
Discussion
The discovery and identification of genomic variants such as SNPs, together with the determination of their location in the genome, can provide valuable information for breeding programs. In plants, many traits of interest have been linked with SNPs23,42,43 and these linkages have been exploited to understand individual variation, community diversity, and the evolution of species44. In this study, we conducted deep Genotyping-by-Sequencing and genome-wide SNP discovery on a diverse collection of accessions representing three different subgenera of the genus Prunus (Amygdalus, Prunus and Cerasus). This included 53 diploid Prunus rootstocks and five scion cultivars. Some of the accessions included in this work are extensively used by the stone fruit industry around the world. In fact, millions of trees on these rootstocks are sold and planted every year and they remain in the orchard for 10–20 years, depending on the fruit species. That makes this research relevant through time and to broad audience, and the results provide valuable insight into genetic diversity and relationships at the interspecific level within Prunus.
SNPs identified from GBS have been previously used in Prunus for phylogenetic and population structure analysis in apricot37 using 90 accessions of different origins and DNA digestion using ApeKI restriction enzyme. Single-digest GBS was also used for phylogenetic analysis using 11 Japanese plum cultivars to explore their natural allelic diversity in relation to the most important phenology events and fruit quality traits38. In this work, we used a double-digest strategy (PstI/MspI) and deep coverage paired-end sequencing for genome-wide representation of each Prunus sample. In our experience and in other published reports, paired-end sequence reads can be more accurately mapped onto the reference genome when compared to single-end reads45, which results in a significantly greater number of quality SNPs and lower amounts of missing data.
The availability of a reference genome sequence allows for a positional reference for each SNP for genome-wide analysis. The group of SNPs identified in the present study could prove useful for various marker-based applications in Prunus. In fact, the genus Prunus is well enabled with the availability of several reference genome sequences25,26,46,47,48,49. However, it must be noted that each genome is prone to harbor insertions and deletions with respect to genomes from other species. For example, in the first work published using GBS, Elshire et al.27 observed that BLAST results indicated that the majority of non-aligning reads represented maize sequences that were absent in the reference genome version used for the analysis. In peach, some genes have been reported as mutated when they were compared with similar sequences from other Prunus species50,51,52,53,54. Nevertheless, in spite of these differences, results obtained in this study further corroborate the high degree of synteny found among Prunus species reported previously34,55,56,57. Even though our analysis included species from three different Prunus subgenera, we observed an exceptionally high read mapping rate to the reference genome.
In comparison to previous studies where genotypes were collected by a community SNP array, only a group of 49 SNPs were in common with the cherry 6 K SNP array v139, and 75 SNPs with the IRSC 9 K peach SNP array v140. This could be explained by technical differences in the two genotyping approaches where peach SNP array does not contain A/T nor C/G SNPs, for example, and because the accessions used for SNP identification are different. Also, GBS allows for discovery of the SNPs that are different between the genotyped samples and the reference genome, therefore reflecting the difference between peach genome sequence and the material analyzed. Among newly discovered SNPs, transitions were more frequent than transversions which was previously observed in different plant species22,58,59,60. This is to be expected because transitions are less likely to result in amino acid substitutions and are therefore more likely to persist as silent substitutions in populations. The Amygdalus subgenus exhibited the lowest percentage of heterozygous SNPs, while Prunus-Amygdalus hybrids had the highest number of heterozygous SNPs. This is logical when the reference genome is considered, where material closer to P. persica is less heterozygous, e.g. ‘Pomona’, a pure P. persica. Other accessions with a low percentage of heterozygous SNPs, ‘Pontaleb’, ‘Mazzard F12/1’ and ‘Bing’, belong to Cerasus subgenus. These results could be explained with variations in the extent of genetic diversity across stone fruit species, ranging from a narrow genetic base in peaches; intermediate for apricots, sweet cherries and sour cherries; and higher genetic variability in almonds and plums61,62.
Classification of 58 Prunus accessions into three major genetically distinct groups was consistent with their expected pedigree relationships and parentage, which is in agreement with previous studies featuring accessions useful as rootstocks for different Prunus species9,18,19,21. Classification of hybrids between Prunus and Amygdalus, e.g. ‘Ishtara’, clearly reflected their origin with the proportion of genetic content belonging to both Prunus and Amygdalus subgenus. An interesting result was observed for Nanking cherry (P. tomentosa), classified as a member of the section Microcerasus within the subgenus Cerasus1 or the subgenus Prunus2,11,63. The P. tomentosa accession was grouped within Prunus in our study, although the structure results reveal similarities with all three subgenera (Amygdalus, Prunus and Cerasus). Mowrey and Werner11 suggested P. tomentosa being more primitive than other Prunus species, which could explain our results.
Detailed analysis of the physical position of each SNP detected in 55 accessions revealed a non-uniform pattern of SNP distribution in all eight peach pseudomolecules, related to gene density along pseudomolecules (Fig. 6). Similar distribution has been observed in Solanum44 using SNP from ESTs of Solanum habrochaites and S. lycopersicum. In the peach genome, most of the regions with low number of SNPs were associated with the putative position of the centromere of each pseudomolecule26. One exception to the SNP density being associated with gene density is the bottom region of pseudomolecule 4, which had high number of SNPs in our material, but low gene density (Figs. 6, 7 and Supplementary Table S4).
The association between single nucleotide change and gene function has been reported for a number of traits44,59,64,65,66,67. Therefore, the identification of non-synonymous SNPs would be biologically meaningful, and useful for functional genomics, molecular genetics, and marker-assisted selection in breeding. Our results revealed that SNPs located in genic regions (90.3% of the total group) were identified in 4,884 genes in the peach genome, with 53% of SNPs located in exons. The number of SNPs located in genic regions is higher than previously observed in two sweet cherry cultivars and their progeny34, where 65.5% of SNPs were located in genic regions and 49.8% were located in exonic regions. Differences could be due to the use of different restriction enzymes, different version of the peach genome (ApeKI and Peach v1.0 in the previous work and PstI/MspI and Peach v2.0 in this work) and material analyzed. Despite the differences in the methodology between the studies, the percentage of SNPs located in exon regions was similar, with less SNPs identified in intergenic regions and higher number of SNPs located in intron and 5′- and 3′-UTR in this study. This information might be useful for future applications because SNPs in the upstream, downstream, and 5′- and 3′- UTR regions might affect transcription and/or translation. However, the actual SNP effects have to be confirmed on case-by-case basis68 because not all mutations are functionally important and different proteins and domains differ in how well they tolerate mutations69.
As expected, the SNPs classified as modifier in our study were more abundant that the other categories, while the high impact SNPs were the smaller proportion. The same was observed in other crops, for example bean70, soybean71 and pear72. In contrast, the SNPs with impact on protein efficiency and loss-of-function, that have a direct impact on gene function with adaptive interference during the course of selection, were reported in a smaller proportion. To the best of our knowledge, this is the first work using members from different subgenera which were analyzed using GBS and where SNPs were classified according to their putative effect on annotated genes. Consequently, it was not possible to compare our results with those from other related works. Similar distribution of SNPs in respect to their putative effect was observed in a collection of bean accessions70 by using the Diversity Arrays Technology methodology (DArT), also based on genome complexity reduction using restriction enzymes (PstI/MseI) and SNP detection through hybridization of PCR fragments73.
The group of 128 high impact SNPs identified in this study, which are located in 119 genes, could have a direct effect on the gene functionality in the group of accessions analyzed. These SNPs caused either stop codon gain, splice donor and acceptor variants or stop codon loss. By performing KOG analysis (Fig. 9 and Supplementary Table S8) of these genes to investigate their putative functional class, it was observed that no functional class could be assigned to a larger fraction (64%) of them and they have been annotated as hypothetical proteins in the peach genome. However, among the genes with predicted class, those involved in metabolism were the most abundant. Our analysis allowed to identify an important group of genes affected in three Prunus subgenera, with nucleotide changes observed for some subgenus and not for others. Detailed analyses are needed, but this information will facilitate investigation of the consequences of predicted SNPs and their biological role. Involvement of SNPs in genes reported to be playing a role in metabolism, cellular processes and signaling, and information storage and processing could be addressed in detail at breeding level.
Conclusion
The 45,382 GBS-derived SNPs identified in this study represent a valuable resource for molecular characterization of commercial and selected Prunus rootstocks. This resource provides foundation for analysis of the genetic diversity among the different interspecific hybrids and species in the germplasm collections of CEAF and EEAD-CSIC for their conservation, management and utilization in current or future rootstock breeding programs. Informative SNPs identified in this study, particularly in coding and non-coding regulatory sequence components of various genes, once validated, can be utilized as potential markers in genetic and association mapping for identifying major trait-regulatory candidate genes/QTLs in Prunus.
Material and Methods
Plant material
The group of 58 diploid accessions used in this study (Table 2) are part of two Prunus rootstock collections: 33 accessions coming from CEAF in Chile and 25 from EEAD-CSIC in Spain. These accessions belong to three Prunus subgenera: Amygdalus (n = 25), Prunus (n = 17), Cerasus (n = 6) and 10 hybrids between subgenera Prunus and Amygdalus. Detailed information about accessions is provided in Table 2. To compare results from GBS, three commercial rootstocks (‘Adara’, ‘Citation’ and ‘Mariana 2624’) were analyzed in duplicate, with one sample from each rootstock collection.
DNA extraction and quantification
For genomic DNA extraction, young leaves of each accession were collected and stored at −80 °C until use. The DNeasy Plant Mini kit (Qiagen) and the NucleoSpin Plant II kit (Macherey-Nagel) were used for DNA extraction for samples from CEAF and EEAD-CSIC, respectively, according to the manufacturer’s instructions. DNA quality was examined by 1% agarose gels and DNA quantity was determined by spectrophotometry (Tecan Tradind AG, Switzerland).
Genotyping-by-Sequencing
GBS was carried out at Clemson University Genomics Computational Laboratory (CUGCL; Clemson, SC, USA). A reduced representation GBS library was prepared using restriction enzymes PstI (methylation sensitive) and MspI (partial sensitivity to methylation), as described by Poland et al.28 in cereals. A total of 200 ng of intact genomic DNA was digested and ligated to custom designed adapter sequences. A total of 58 GBS libraries were sequenced on an Illumina HiSeq2500 using a 2x125 bp paired-end read module across 2 high-output lanes. Raw sequence data was demultiplexed and preprocessed for errors using the Stacks demultiplex tool74. Sample specific sequences were aligned to the eight pseudomolecules representing the eight chromosomes of the peach genome assembly (Peach v2.0)26 with the GMAP/GSNAP release 816.1675. The resulting variant call file (.vcf) was filtered for SNPs with a minimum depth (DP) of six, and present in at least 80% of the accessions. Mean coverage of each GBS SNP was determined by creating a.BED file from the final SNP set and generating a bed graph with the genomecov function of bedtools v. 2.28.076, and intersecting the bedgraph with the SNP.bed file with the intersect function in bedtools. The mean coverage of each sample was determined with in house scripts. SNPs were extracted using the GBS pipeline implemented in TASSEL 5.2.5 software77 and accessions were called using minor allele frequency (MAF) > 0.05.
SNPs analysis
SNPs were labeled according to the pseudomolecules in the peach genome (Pp01 to Pp08), followed by the physical position in base pairs (bp). Location of each SNP within genic [exonic, intronic, and untranslated regions (UTR)] and intergenic regions was determined using a custom Perl script (www.perl.org) with Peach v2.0 as reference. The physical position of each SNP was used to identify common markers among this study, the RosBREED cherry 6 K SNP array v139 and the IRSC 9 K peach SNP array v140.
Transitions/transversions and percentage of heterozygous positions were determined using SNiPlay3 (http://sniplay.southgreen.fr)78.
Phylogenetic and population structure analysis
An UPGMA dendrogram was constructed using Archeopteryx software within TASSEL77. In order to identify population structure, the SNP genotyping information was analyzed with the program STRUCTURE v2.3.479. Analysis were carried out for a range of K values from 1 to 10, with 10 runs for each K. A burn-in of 5,000 and 50,000 MCMC replications were implemented for each run. The optimal number of K clusters was estimated using the ΔK parameter of Evanno et al.79 in Structure Harvester (http://taylor0.biology.ucla.edu/structureHarvester/)80. Accessions were subdivided into different populations according to their maximum membership probability among the populations and the membership probabilities threshold of 0.80. Furthermore, principal components analysis (PCA) was performed on genotype scores using the PCA function in TASSEL and visualized using Infostat v201781 to confirm population structure among accessions.
Functional characterization of SNPs
Circos software v 0.69–382 was used to plot the histograms of both gene and SNP density for each pseudomolecule of the peach genome sequence26. Gene and SNP density were assessed and plotted in a window of 1000/kb. In a first analysis, SNP density was determined for 55 accessions (considering only one of the replicates of ‘Adara’, ‘Citation’ and ‘Mariana 2624’). In a second analysis, accessions were separated by subgenus (25 accessions from Amygdalus subgenus, 15 from Prunus and 5 from Cerasus; Table 2). Prunus-Amygdalus hybrids and Nanking cherry were just considered for the first analysis of 55 accessions. A Chi-square test was performed to compare the SNP distribution in genic (exonic, intronic and 5′- and 3′-UTR) and intergenic regions for the three subgenus (Amygdalus, Prunus and Cerasus) and for the group of 55 accessions analyzed in this study.
Prediction of SNP effects was performed using SnpEff v 4.3e68 based on the P. persica gene annotation (www.rosaceae.org). Whenever multiple transcripts for a gene exist, the effect on each transcript was analyzed. The SNP predicted effects were categorized by impact, as modifier (with impact on noncoding regions), low (synonymous substitution); moderate (non-synonymous substitution); or high (disruptive impact on the protein). To investigate the putative function of the genes containing high impact SNPs, a eukaryotic orthologous group (KOG) analysis was carried out using tools from Join Genome Institute (JGI, https://jgi.doe.gov).
Data availability
The dataset generated for this study is available in the NCBI-SRA database, BioProject number PRJNA489327.
References
Rehder, A. A manual of cultivated trees and shrubs hardy in North America exclusive of the Subtropical and warmer temperate regions, 2nd ed. MacMillan, New York, USA, pp. 162. (1940).
Aradhya, M. K., Weeks, C. & Simon, C. J. Molecular characterization of variability and relationships among seven cultivated and selected wild species of Prunus L. using amplified fragment length polymorphism. Sci. Hortic. 103(1), 131–144, https://doi.org/10.1016/j.scienta.2004.05.004 (2004).
Dozier, W. A., Knowles, J. W. & Carlton, C. C. Survival, growth, and yield of peach trees as affected by rootstocks. HortScience 19, 26–30 (1984).
Zarrouk, O., Gogorcena, Y., Gómez-Aparisi, J., Betrán, J. A. & Moreno, M. A. Influence of peach x almond hybrids rootstocks on flower and leaf mineral concentration, yield and vigour of two peach cultivars. Sci. Hortic. 106, 502–514, https://doi.org/10.1016/j.scienta.2005.04.011 (2005).
Jiménez, S., Pinochet, J., Gogorcena, Y., Betrán, J. A. & Moreno, M. A. Influence of different vigour cherry rootstocks on leaves and shoots mineral composition. Sci. Hortic. 112(1), 73–79, https://doi.org/10.1016/j.scienta.2006.12.010 (2007).
Mestre, L., Reig, G., Betrán, J. A., Pinochet, J. & Moreno, M. A. Influence of peach-almond hybrids and plum-based rootstocks on mineral nutrition and yield characteristics of ‘Big Top’ nectarine in replant and heavy-calcareous soil conditions. Sci. Hort. 192, 475–481, https://doi.org/10.1016/j.scienta.2015.05.020 (2015).
Moreno, M. A. Breeding and selection of Prunus rootstocks at the Aula Dei Experimental station, Zaragoza, Spain. Acta Hort. 658, 519–528 (2004).
Reig, G. et al. Long-term graft compatibility study of peach-almond hybrid and plum based rootstocks budded with European and Japanese plums. Sci. Hortic. 243, 392–400, https://doi.org/10.1016/j.scienta.2018.08.038 (2019).
Bouhadida, M. et al. Molecular characterization and genetic diversity of Prunus rootstocks. Sci. Hortic. 120, 237–245, https://doi.org/10.1016/j.scienta.2008.11.015 (2009).
Font i Forcada, C. et al. Sugars and organic acids profile and antioxidant compounds of nectarine fruits influenced by different rootstocks. Sci. Hortic. 248, 145–153, https://doi.org/10.1016/j.scienta.2018.12.010 (2019a).
Mowrey, B. D. & Werner, D. J. Phylogenetic relationships among species of Prunus as inferred by isozyme markers. Theor. Appl. Genet. 80, 129 (1990).
Uematsu, C., Sasakuma, T. & Ogihara, Y. Phylogenetic relationships in the stone fruit group of Prunus as revealed by restriction fragment analysis of chloroplast DNA. Jpn. J. Genet. 66, 59–69 (1991).
Badenes, M. L. & Parfitt, D. E. Phylogenetic relationships of cultivated species from an analysis of chloroplast DNA variation. Theor. Appl. Genet. 90, 1035–1041, https://doi.org/10.1007/BF00222918 (1995).
Bouhadida, M. et al. Chloroplast DNA diversity in Prunus and its implication on genetic relationships. J. Am. Soc. Hort. Sci. 132(5), 670–679, https://doi.org/10.21273/JASHS.132.5.670 (2007).
Lee, S. & Wen, J. A phylogenetic analysis of Prunus and the Amygdaloideae (Rosaceae) using ITS sequences of nuclear ribosomal DNA. Am. J. Bot. 88, 150–160 (2001).
Bortiri, E. et al. Phylogeny and systematics of Prunus (Rosaceae) as determined by sequence analysis of ITS and the chloroplast trnL–trnF spacer DNA. Syst. Bot. 26, 797–807, https://doi.org/10.1043/0363-6445-26.4.797 (2001).
Lu, Z.-X., Reighard, G. L., Baird, W. V., Abbott, A. G. & Rajapakse, S. Identification of peach rootstock cultivars by RAPD markers. HortScience 31, 127–129 (1996).
Casas, A. M., Igartua, E., Balaguer, G. & Moreno, M. A. Genetic diversity of Prunus rootstocks analysed by RAPD markers. Euphytica 110, 139–149, https://doi.org/10.1023/A:100374531 (1999).
Serrano, B., Gómez-Aparisi, J. & Hormaza, J. I. Molecular fingerprinting of Prunus rootstocks using SSRs. J. Hortic. Sci. Biotech. 77, 368–372, https://doi.org/10.1080/14620316.2002.11511507 (2002).
Liu, X., Reighard, G. L., Swire-Clark, G. A. & Baird, W. V. Peach rootstock identification by DNA-fingerprinting with microsatellite (SSR) markers. J. Am. Pomol. Soc. 61, 162–166 (2007).
Arismendi, M. J. et al. Characterization of genetic diversity of stone fruit rootstocks used in Chile by means of microsatellite markers. J. Amer. Soc. Hort. Sci. 137(5), 302–310, https://doi.org/10.21273/JASHS.137.5.302 (2012).
Aranzana, M. J., Illa, E., Howad, W. & Arús, P. A first insight into peach [Prunus persica (L.) Batsch] SNP variability. Tree Genet. Genom. 8(6), 1359–1369, https://doi.org/10.1007/s11295-012-0523-6 (2012).
Mammadov, J., Aggarwal, R., Buyyarapu, R. & Kumpatla, S. SNP markers and their impact on plant breeding. Int. J. Plant Genomics 2012, 1–11, https://doi.org/10.1155/2012/728398 (2012).
Hirakawa, H. et al. Genome-wide SNP genotyping to infer the effects on gene functions in tomato. DNA Res. 20(3), 221–233, https://doi.org/10.1093/dnares/dst005 (2013).
Verde, I. et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nature Genet. 45, 487–494, https://doi.org/10.1038/ng.2586 (2013).
Verde, I. et al. The Peach v2.0 release: high-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genomics 18, 225, https://doi.org/10.1186/s12864-017-3606-9 (2017).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS ONE 6(5), e19379, https://doi.org/10.1371/journal.pone.0019379 (2011).
Poland, J. A., Brown, P. J., Sorrells, M. E. & Jannink, J. L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PloS ONE 7(2), e32253, https://doi.org/10.1371/journal.pone.0032253 (2012).
Narum, S. R., Buerkle, C. A., Davey, J. W., Miller, M. R. & Hohenlohe, P. A. Genotyping by sequencing in ecological and conservation genomics. Mol. Ecol. 22(11), 2841–2847, https://doi.org/10.1111/mec.12350 (2013).
Kim, C. et al. Application of genotyping by sequencing technology to a variety of crop breeding programs. Plant Sci. 242, 14–22, https://doi.org/10.1016/j.plantsci.2015.04.016 (2016).
Yao, X. et al. Construction of a high-density genetic map: genotyping by sequencing (GBS) to map purple seed coat color (Psc) in hulless barley. Hereditas 155, 37, https://doi.org/10.1186/s41065-018-0072-6 (2018).
Bielenberg, D. G. et al. Genotyping by sequencing for SNP-based linkage map construction and QTL analysis of chilling requirement and bloom date in peach [Prunus persica (L.) Batsch]. PLoS ONE 10(10), e0139406, https://doi.org/10.1371/journal.pone.0139406 (2015).
Goonetilleke, S. N. et al. Genotyping by Sequencing in almond: SNP. discovery, linkage mapping, and marker design. G3 (Bethesda) 8(1), 161–172, https://doi.org/10.1534/g3.117.300376 (2018).
Guajardo, V. et al. Construction of high density sweet cherry (Prunus avium L.) linkage maps using microsatellite markers and SNPs detected by genotyping-by-sequencing (GBS). PLoS ONE 10(5), e0127750, https://doi.org/10.1371/journal.pone.0127750 (2015).
Nuñez-Lillo, G. et al. Identification of candidate genes associated with mealiness and maturity date in peach [Prunus persica (L.) Batsch] using QTL analysis and deep sequencing. Tree Genet. Genomes 11(4), 86, https://doi.org/10.1007/s11295-015-0911-9 (2015).
González, M. et al. Genetic structure based on EST–SSR: a putative tool for fruit color selection in Japanese plum (Prunus salicina L.) breeding programs. Mol. Breed. 36(6), 1–15, https://doi.org/10.1007/s11032-016-0491-x (2016).
Gürcan, K., Teber, S., Ercisli, S. & Yilmaz, K. U. Genotyping by sequencing (GBS) in apricots and genetic diversity assessment with GBS-derived single-nucleotide polymorphisms (SNPs). Biochem. Genet. 54(6), 854–885, https://doi.org/10.1007/s10528-016-9762-9 (2016).
Salazar, J. A. et al. Development and applicability of GBS approach for genomic studies in Japanese plum (Prunus salicina Lindl.). J. Hortic. Sci. Biotech. 94(3), 284–294, https://doi.org/10.1080/14620316.2018.1543559 (2019).
Peace, C. et al. Development and evaluation of a genome-wide 6K SNP array for diploid sweet cherry and tetraploid sour cherry. PLoS ONE 7(12), e48305, https://doi.org/10.1371/journal.pone.0048305 (2012).
Verde, I. et al. Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PloS ONE 7(4), e35668, https://doi.org/10.1371/journal.pone.0035668 (2012).
Campoy, J. A. et al. Genetic diversity, linkage disequilibrium, population structure and construction of a core collection of Prunus avium L. landraces and bred cultivars. BMC Plant Biol. 16(1), 49, https://doi.org/10.1186/s12870-016-0712-9 (2016).
Huq, M. A. et al. Identification of functional SNPs in genes and their effects on plant phenotypes. J. Plant. Biotechnol. 43 (1), 1–11, https://doi.org/JPB-43-001 (2016).
Zhang, W., Mirlohi, S., Li, X. & He, Y. Identification of functional single-nucleotide polymorphisms affecting leaf hair number in Brassica rapa. Plant Physiol. 177(2), 490–503, https://doi.org/10.1104/pp.18.00025 (2018).
Bhardwaj, A., Dhar, Y. V., Asif, M. H. & Bag, S. K. In silico identification of SNP diversity in cultivated and wild tomato species: insight from molecular simulations. Sci. Rep. 6, 38715, https://doi.org/10.1038/srep38715 (2016).
Shirasawa, K., Hirakawa, H. & Isobe, S. Analytical workflow of double-digest restriction site-associated DNA sequencing based on empirical and in silico optimization in tomato. DNA Res. 23(2), 145–153, https://doi.org/10.1093/dnares/dsw004 (2016).
Zhang, Q. et al. The genome of Prunus mume. Nat. Commun. 3, 1318, https://doi.org/10.1038/ncomms2290 (2012).
Shirasawa, K. et al. The genome sequence of sweet cherry (Prunus avium) for use in genomics-assisted breeding. DNA Res. 24, 499–508, https://doi.org/10.1093/dnares/dsx020 (2017).
Baek, S. et al. Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol. 19(1), 127, https://doi.org/10.1186/s13059-018-1497-y (2018).
Sánchez-Pérez, R. et al. Mutation of a bHLH transcription factor allowed almond domestication. Science 364(6445), 1095–1098, https://doi.org/10.1126/science.aav8197 (2019).
Bielenberg, D. G. et al. A deletion affecting several gene candidates is present in the evergrowing peach mutant. J. Hered. 95(5), 436–444, https://doi.org/10.1093/jhered/esh057 (2004).
Tao, R. et al. Self-compatible peach (Prunus persica) has mutant versions of the S haplotypes found in self-incompatible Prunus species. Plant Mol. Biol. 63(1), 109–123, https://doi.org/10.1007/s11103-006-9076-0 (2007).
Vendramin, E. et al. A unique mutation in a MYB gene cosegregates with the nectarine phenotype in peach. PLoS ONE 9(3), e90574, https://doi.org/10.1371/journal.pone.0090574 (2014).
Cheng, J. et al. A small indel mutation in an anthocyanin transporter causes variegated colouration of peach flowers. J. Exp. Bot. 66(22), 7227–7239, https://doi.org/10.1093/jxb/erv419 (2015).
López-Girona et al. A deletion affecting an LRR-RLK gene co-segregates with the fruit flat shape trait in peach. Sci. Rep. 7(1), 6714, https://doi.org/10.1038/s41598-017-07022-0 (2017).
Dirlewanger, E. et al. Comparative mapping and marker assisted selection in Rosaceae fruit crops. Proc. Natl. Acad. Sci. USA 101, 9891–9896, https://doi.org/10.1073/pnas.0307937101 (2004).
Olmstead, J. et al. Construction of an intra-specific sweet cherry (Prunus avium L.) genetic linkage map and synteny analysis with the Prunus reference map. Tree Genet. Genomes 4, 897–910, https://doi.org/10.1007/s11295-008-0161-1 (2008).
Cabrera, A. et al. Rosaceae conserved orthologous sequences marker polymorphism in sweet cherry germplasm and construction of a SNP-based map. Tree Genet. Genomes 8(2), 237–247, https://doi.org/10.1007/s11295-011-0436-9 (2012).
Bajaj, D. et al. Genome-wide high-throughput SNP discovery and genotyping for understanding natural (functional) allelic diversity and domestication patterns in wild chickpea. Sci. Rep. 5, 12468, https://doi.org/10.1038/srep12468 (2015).
Kujur, A. et al. Employing genome-wide SNP discovery and genotyping strategy to extrapolate the natural allelic diversity and domestication patterns in chickpea. Front. Plant Sci. 6, 162, https://doi.org/10.3389/fpls.2015.00162 (2015).
Ariani, A. Mier y Teran, J. C. B. & Gepts, P. Genome-wide identification of SNPs and copy number variation in common bean (Phaseolus vulgaris L.) using genotyping-by-sequencing (GBS). Mol. Breed. 36(7), 1–11, https://doi.org/10.1007/s11032-016-0512-9 (2016).
Byrne, D. H. Isozyme variability in four diploid stone fruits compared with other woody perennial plants. J. Hered. 81, 68–71, https://doi.org/10.1093/oxfordjournals.jhered.a110927 (1990).
Granger, A. R., Clark, G. R. & Jackson, J. F. Sweet cherry cultivar identification by leaf isozyme polymorphism. Theor. Appl. Genet. 86(4), 458–464, https://doi.org/10.1007/BF00838561 (1993).
Bortiri, E., Oh, S.-H., Gao, F.-Y. & Potter, D. The phylogenetic utility of nucleotide sequences of sorbitol 6-phosphate dehydrogenase in Prunus (Rosaceae). Am. J. Bot. 89(10), 1697–1708, https://doi.org/10.3732/ajb.89.10.1697 (2002).
Shirasawa, K. et al. Genome-wide association studies using single nucleotide polymorphism markers developed by re-sequencing of the genomes of cultivated tomato. DNA Res. 20(6), 593–603, https://doi.org/10.1093/dnares/dst033 (2013).
Cao, K. et al. Genome-wide association study of 12 agronomic traits in peach. Nat. Commun. 7, 13246, https://doi.org/10.1038/ncomms13246 (2016).
Urrestarazu, J. et al. Genome-wide association mapping of flowering and ripening periods in apple. Front. Plant Sci. 8, 1923, https://doi.org/10.3389/fpls.2017.01923 (2017).
Font i. Forcada, C., Guajardo, V., Reyes Chin Wo, S. & Moreno, M. Á. Association mapping analysis for fruit quality traits in Prunus persica using SNP markers. Front. Plant Sci. 9, 2005, https://doi.org/10.3389/fpls.2018.02005 (2019b).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6(2), 80–92, https://doi.org/10.4161/fly.19695 (2012).
Yates, C. M. & Sternberg, M. J. Proteins and domains vary in their tolerance of non-synonymous single nucleotide polymorphisms (nsSNPs). J. Mol. Biol. 425(8), 1274–1286, https://doi.org/10.1016/j.jmb.2013.01.026 (2013).
Valdisser, P. A. et al. In-depth genome characterization of a Brazilian common bean core collection using DArTseq high-density SNP genotyping. BMC Genomics 18(1), 423, https://doi.org/10.1186/s12864-017-3805-4 (2017).
Ramakrishna, G. et al. Genome-wide identification and characterization of InDels and SNPs in Glycine max and Glycine soja for contrasting seed permeability traits. BMC Plant Biol. 18(1), 141, https://doi.org/10.1186/s12870-018-1341-2 (2018).
Montanari, S. et al. Development of a highly efficient Axiom™ 70 K SNP array for Pyrus and evaluation for high-density mapping and germplasm characterization. BMC Genomics 20(1), 331, https://doi.org/10.1186/s12864-019-5712-3 (2019).
Jaccoud, D., Peng, K., Feinstein, D. & Kilian, A. Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res. 29(4), e25, https://doi.org/10.1093/nar/29.4.e25 (2001).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140, https://doi.org/10.1111/mec.12354 (2013).
Wu, T. D., Reeder, J., Lawrence, M., Becker, G. & Brauer, M. J. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. In: Mathé E., Davis S. (eds) Statistical Genomics. Methods in Molecular Biology, vol 1418. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3578-9_15 (2016).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6), 841–842, https://doi.org/10.1093/bioinformatics/btq033 (2010).
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635, https://doi.org/10.1093/bioinformatics/btm308 (2007).
Dereeper, A., Nicolas, S. & Le Cunff, L. SNiPlay: a web-based tool for detection, management and analysis of SNPs. Application to grapevine diversity projects. BMC Bioinformatics 12(1), 134, https://doi.org/10.1186/1471-2105-12-134 (2011).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620, https://doi.org/10.1111/j.1365-294X.2005.02553.x (2005).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4(2), 359–361, https://doi.org/10.1007/s12686-011-9548-7 (2012).
Di Rienzo, J. A. et al. InfoStat versión Grupo InfoStat, FCA, Universidad Nacional de Córdoba, Argentina. http://www.infostat.com.ar (2011).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19(9), 1639–1645, https://doi.org/10.1101/gr.092759.109 (2009).
Acknowledgements
This research was funded by CONICYT-REGIONAL/GORE O’HIGGINS/CEAF/R19A10003; FONDECYT 3160316; FONDECYT 1160706; CONICYT R16F20006; and the Spanish Ministry of Science, Innovation and Universities (MICINN) grants RFP2015-00019 and RTI2018-094176-R-C33, cofunded by FEDER. The authors are grateful to Dr. Ana María Casas and Dr. María Pilar Vallés at EEAD-CSIC, Spain, for helpful discussions and providing equipment access; Dr. Michelle Morales from CEAF, Chile, for her valuable assistance in PCA analysis, and Francisco Correa from INIA Rayentué-CEAF, Chile, for his valuable assistance in bioinformatics analysis.
Author information
Authors and Affiliations
Contributions
V.G., R.A. and M.Á.M. conceived and designed the experiments. V.G. and S.S. performed the experiments. V.G., S.S., C.S. and K.G. analyzed the data. R.A., C.S., K.G. and M.Á.M.: contributed with reagents, materials and analysis tools. All authors contributed to the manuscript preparation, and read and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guajardo, V., Solís, S., Almada, R. et al. Genome-wide SNP identification in Prunus rootstocks germplasm collections using Genotyping-by-Sequencing: phylogenetic analysis, distribution of SNPs and prediction of their effect on gene function. Sci Rep 10, 1467 (2020). https://doi.org/10.1038/s41598-020-58271-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58271-5
This article is cited by
-
Single nucleotide polymorphisms reveal the uniqueness of Gushan semi-rock tea in the tea germplasm resources of Fujian, China
Genetic Resources and Crop Evolution (2023)
-
Genome resequencing reveals the population structure and genetic diversity of almond in Xinjiang, China
Genetic Resources and Crop Evolution (2023)
-
Fine-tuning the performance of ddRAD-seq in the peach genome
Scientific Reports (2021)
-
Genetic diversity and population structure of advanced clones selected over forty years by a potato breeding program in the USA
Scientific Reports (2021)
-
Genetic diversity of Norway spruce ecotypes assessed by GBS-derived SNPs
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
