
Abstract
In paper industry, xylanases are used to increase the pulp properties in bleaching process as its eco-friendly nature. The xylanases activity is hindered by high temperature and alkaline conditions with high enzyme production cost in the paper industry. Here, XynHB, an alkaline stable xylanase from Bacillus pumilus HBP8 was mutated at N188A to XynHBN188A. Expressed mutant in E. coli showed 1.5-fold higher xylanase activity than XynHB at 60 °C. The mutant expressed in Pichia pastoris was glycosylated, remained stable for 30 min at 60 °C. XynHBN188A optimized based on codon usage bias for P. pastoris (xynHBN188As) showed an increase of 39.5% enzyme activity. The strain Y16 forming the largest hydrolysis halo in the xylan plate was used in shake flask experiments produced an enzyme activity of 6,403 U/ml. The Y16 strain had 9 copies of the recombinant xynHBN188As gene in the genome revealed by qPCR. The enzymatic activity increased to 48,241 U/ml in a 5 L fermentor. Supplement of 15 U/g xylanase enhanced the brightness of paper products by 2% in bleaching experiment, and thereby improved the tensile strength and burst factor by 13% and 6.5%, respectively. XynHBN188As has a great potential in paper industries.
Similar content being viewed by others

Introduction
Paper industry is one of the fastest growing sectors. The chemicals used in bleaching process are large amounts of chlorinated inorganic compounds, which are toxic, mutagenic, and carcinogenic in nature1. Under these circumstances, xylanase is used in the paper industry to reduce the harmful effects of chemicals causing adverse effect. The positive effect of xylanase generally attributes to the breaking down of the xylan, thereby breaking down the link between the cellulose and lignin. Once the lignin is detached from cellulose, it is readily reduced in the subsequent bleaching steps. In addition, after kraft pulping, xylan generally co-crystalize on the surface of the fiber, causing a physical barrier for the reagents to flow into the fiber2,3. Therefore, the degradation of xylan would facilitate the influx of subsequent bleaching agents. It’s known that the use of xylanase could reduce the damage to pulp fibers, and also create high quality rayon grade and superior quality dissolving pulps, providing an alternative and cost-effective method for the bleaching process3.
However, the high temperature and alkaline conditions influence the use of different xylanases in paper and pulp industry. Most of the presently used xylanases doesn’t withstand the high temperature or the alkaline condition causing reduced activity, which doesn’t fit to the industrial standards, so there is a great need for the thermostable xylanases effective at alkaline pH with higher activity. Xylanases were mainly classified into two families, i.e. glycosyl hydrolase GH 10 and GH 11. The GH10 families namely, endo-β-1, 4-D xylanase have a higher molecular weight (≥30 kDa), low isoelectric point, but studies have shown that these enzymes are not substrate specific. The GH11 families are the smallest xylanases (generally less than 30 kDa), which penetrate into the pulp easily with high specificity, but do not tolerate high temperature4. Previously, many efforts have been reported to improve the thermo-stability of xylanases by different strategies5,6. N-terminal region of xylanase was reported to play a crucial role in its thermo-stability, so single residue substitutions and chimeric alterations in the vicinity of the N-terminal region of the protein make a big difference7,8,9. Thermo-stability of xylanases was improved by the introduction of disulphide bridges, thereby enhancing the protein positive charges10,11,12,13,14. Also, increasing the stability of a protein by decreasing the configuration entropy of unfolding, such as the hydrogen bond network, can provide significant energetic contribution to the thermo-stability of a protein15,16. Deletion of the carbohydrate-binding modules (CBM) has effect on the thermo-stability of GH10 and GH11 xylanases. The truncated mutant XynATM1 harboring a GH11 catalytic module without CBM showed an improved thermal stability compared to XynA17. Moreover, DNA shuffling and error prone PCR were applied to engineer recombinant xylanase with improved thermo-stability18,19,20. These findings prompted the engineering of xylanase for large scale industrial application in paper industry, but further work is needed to develop a highly thermo-stable xylanase having better enzyme activity in alkaline condition.
The recombinant strains producing high-yield of xylanase will be better for industrial application with less downstream purification, so xylanase production will be cheaper and cost efficient21,22. Several studies have reported the role of different host strains produce high level of exogenous xylanase, like Bacillus, Trichoderma reesei, Pichia pastoris, Saccharomyces cerevisiae and Escherichia coli23,24,25,26,27. But, P. pastoris is known for its properties of efficient enzyme secretion, cellulase free, and fast growth with high cell density in simple media28. Some GH10 xylanases have been successfully expressed in P. pastoris, which is thermophilic but can be suitable for acidic conditions only and they showed the highest xylanase activity of 73,400 U/ml29.
In our previous study, the GH11 xylanase gene xynHB was cloned from Bacillus pumilus HBP8 and expressed in P. pastoris. The xylanase activity in the supernatant of the recombinant P. pastoris expressing xynHB was 644 U/ml30, and recombinant XynHB was effective in alkaline condition (pH 8.6) with 90% activity. However, the thermo-stability and expression yield did not meet the industrial standards for paper industry, since the optimal temperature of this recombinant xylanase was 40–60 °C, with only 20% residual activity after 30 min preincubation at 60 °C. In the present study, a site mutagenesis was performed to improve thermo-stability of recombinant XynHB as well as better protein expression level in P. pastoris by gene codon usage optimization and multiple gene insertion.
Methods
Strains, plasmids and chemicals
E. coli DH5α, E. coli BL21(DE3) were purchased from TransGen Biotech (Beijing, China). P. pastoris GS115 were purchased from Invitrogen (USA). The expression vector pET28a(+) of E. coli was purchased from Novagen (Germany). The recombinant plasmid pHBM130 encoding xynHB gene expressing xylanase30 and the expression vector pHBM905A of P. pastoris31 was constructed previously.
Synthesis of PCR primers and DNA sequencing were performed by GenScript Co. Ltd (Nanjing, China). Restriction enzymes, Ex Taq DNA Polymerase, LA DNA Polymerase and T4 DNA Ligase were purchased from TakaRa (Dalian, China). All chemicals were of analytical grade and obtained from commercial suppliers.
For culturing different recombinants of P. pastoris, Buffered Glycerol-complex Medium (BMGY), Buffered Methanol-complex Medium (BMMY), Minimal Dextrose Medium (MD) and basal salt medium (BSM) were prepared as described in previous study32.
Construction of the E. coli expression plasmid and site-directed mutagenesis of xynHB
The target gene xynHB (GenBank accession number AY954630) was amplified from plasmid pHBM130 by polymerase chain reaction (PCR) using primers HB1 and HB2 (with BamHI and SalI site) (Table 1). Gene amplification was performed in a 50 μL PCR volume containing 10 ng of plasmid pHBM130, 0.5 μM of primer pairs, 5 μL of 10 × Ex Taq Buffer, 200 μM dNTPs and 1 U of Ex Taq DNA Polymerase (Takara). Amplification was performed in a Bio-rad T100 Thermal Cycler (USA) with 25 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min. The PCR products were digested by BamHI/SalI and ligated into BamHI/SalI digested vector pET28a and transformed into E. coli DH5α. The recombinant plasmid pET28a-xynHB was confirmed by PCR and restriction enzymes digestion.
Site-directed mutagenesis was performed by the overlapping extension method using plasmid pET28a-xynHB as a template, and N188AF and N188AR (Table 1) as primers. PCR was performed with LA DNA Polymerase (TaKaRa, China). The purified PCR products were digested by DpnI, and transformed into E. coli DH5α. The mutants were screened on LB plates supplemented with 50 μg/mL of kanamycin and further verified by gene sequencing.
Expression and purification of the XynHB and XynHBN188A in E. coli
Recombinant plasmid was purified and transformed into E. coli BL21 (DE3) followed by culturing in LB-kanamycin (50 μg/ml) medium at 37 °C and 200 rpm until the OD600 reached 0.6. The expression of xylanase was induced with isopropylthiogalactoside (IPTG, 0.5 mM) for continuing 12 h cultivation at 18 °C. Cells were harvested by centrifugation followed by ultrasonication. His • Bind® Kits (Novagen) were used according to the manufacturer’s instructions to purify xylanase. The molecular weight and homogeneity of the protein were evaluated by SDS-PAGE, followed by Coomassie Brilliant Blue G-250 staining33.
Determination of xylanase activity and its thermo-stability
Xylanase were assayed by measuring the reducing sugar released from beechwood xylan by dinitrosalicylic acid method. Xylanase activity was assayed by incubating 1 mL of enzymatic extract with 1 mL of 1% beechwood xylan solution (Sigma, USA) in 50 mM Tris-HCI buffer pH 8.0 as the substrate. The mixture was incubated at 50 °C for 10 min. One unit of enzyme activity was defined as the amount of enzyme capable of releasing 1 μmol of reducing sugar from xylan per minute. Xylose was used as a standard. The protein concentration was determined by Bradford assay using bovine serum albumin (BSA) as the standard. To investigate the thermo-stability, the purified enzyme was incubated at 60 °C. Residual xylanase activity was determined at regular time intervals of 5 min in 50 °C and pH 8.0 for 10 min. The residual activity (%) was expressed as a ratio in the percentage of untreated xylanase activity. All experiments were performed in triplicate.
The analysis of structure variation of XynHB and XynHBN188A
To investigate the structural changes responsible for the improved stability of XynHBN188A, the 3-D structure of XynHB and XynHBN188A were simulated by using MODELLER34. The template search for homology modeling was performed by full multiple alignments and searching in PDB. In our results, a GH11 xylanase, with its PDB number of 1IGO, was identified as the template. XynHB shared 85% amino acid sequence identity with 1IGO. Then, the structure comparison was performed by Discovery Studio (DS) client software (Accelrys Inc., San Diego, CA). The amino acids from 35 to 40 in the N-terminus were selected for the analysis of classical hydrogen bonds.
Codon optimization of xynHBN188A and expression in P. pastoris
In order to further increase the expression level of xylanase in P. pastoris, the gene sequence of xynHBN188A was optimized based on the codon usage bias of P. pastoris. The codon-optimized gene was designed by DNAworks (http://helixweb.nih.gov/dnaworks/) and synthesized by Genscript (China). Primers HBsF and HBsR (Table 1) were used to amplify xynHBN188As gene and HB3 and HB4 primers (Table 1) for xynHBN188A. The partial CpoI and NotI (TaKaRa) are underlined. The PCR products were treated with T4 DNA Polymerase and 1 mM dTTP for 20 min at 12 °C for overhangs. The expression vector of P. pastoris pHBM905A with 5′AOX1 gene promoter was digested by CpoI and NotI restriction enzymes35. The treated PCR products were ligated with pHBM905A downstream of the MFα secretion signal sequence. The ligation products were transformed into E. coli DH5α. The transformants were screened on LB plates (100 μg/mL ampicillin), and further confirmed by colony PCR and DNA sequencing.
The recombinant plasmids (Figure S1) were linearized using SalI and transformed into P. pastoris GS115 by electroporation (10000 V/cm, 4 ms, Bio-Rad MicroPulser Electroporator, USA). Transformants were selected on histidine-deficient MD plates and incubated at 30 °C for 3 days. Positive transformants were identified on an upside down BMMY plate containing 0.5% Remazol Brilliant Blue (RBB)-xylan with methanol dropping uniformly on the petri dish cover at 28 °C. Three colonies with the minimum halo were selected for shake flask fermentation as described in previous report with minor modification31. Cells were cultured in 25 ml of BMGY medium for 48 h and harvested. They were transferred into 25 ml of BMMY medium to induce xylanase expression and measured at regular intervals. Three replicates were performed for each transformant to test the activity.
Screening of genetic-engineering yeast with high-yield XynHBN188A
Thousands of transformants were inoculated on the BMMY plates containing 0.5% (w/v) RBB-xylan, and the inducing method was described previously in the Methods 2.6. The colonies with the largest halo on each plate were inoculated on another RBB-xylan BMMY plate to perform the second level of screening to identify colonies having large halo. The colony with the largest halo was selected for shake flask fermentation.
Determination of the copy number of xynHBN188As in the genome
Total DNA was isolated from P. pastoris according to the method of Hoffman and Winston36. A modified quantitative real-time PCR method was used to determine copy numbers of the target gene in cells. Glyceraldehydes-3-phosphate dehydrogenase (GAP) gene of P. pastoris was used as the reference gene. The HB3S and HB4S primers (Table 1) were used to amplify xynHBN188As and the GAPF and GAPR primers (Table 1) were used to amplify GAP. Strain P81 was used as a calibrator sample since it displayed the smallest halo on the RBB-xylan plate with minimum xylanase activity, so it is assumed that it possessed a single copy xylanase gene.
Real-time PCR (qPCR) was performed in the MiniOpticon™ Real-Time PCR System (Bio-Rad, Hercules, CA) using SYBR® Premix Ex Taq™ (TliRNaseH Plus) (Takara, China). The PCR reaction mixture contained 10 ng of template DNA, 0.5 μM of primer pairs, 12.5 μL of the SYBR® Premix Ex Taq™ and 25 μL sterilized distilled water. The PCR assay included an initial denaturation step at 95 °C for 5 min, followed by 40 cycles of 30 s at 95 °C, 30 s at 60 °C, and 20 s at 72 °C. Fluorescent signal measurements were carried out during the elongation step. The experiment was repeated at two independent times. The copy numbers of the xynHBN188As gene integrated in the genome of recombinant P. pastoris was calculated by the ratio of the copy numbers of the target gene against GAP using the ΔΔCT method of relative quantification37.
High-density fermentation of the recombinant yeast strain
The fermentation was carried out in a 5 L fermentor with 2 L BSM supplemented containing 8 mL/L PTM1. High cell density fermentation of P. pastoris was applied based on the Pichia Fermentation Process Guidelines (Invitrogen). The pH and the temperature were set at 6 and 28 °C, respectively. During the cell growth phase, the cells were grown until the glycerol were exhausted, which indicates the increase in dissolved oxygen (DO) level. When the glycerol was exhausted around 12 h, the feeding medium containing 50% (w/v) glycerol and 12 mL/L PTM1 solution was pumped in according to a predetermined DO level (10–20%). When the OD600 reached 360, the glycerol feed was stopped, which raised DO after about 30 min. At that time, pure methanol containing 12 mL/L PTM1 solution was fed to induce the targeted gene expression, and the fermentation temperature were adjusted to 25 °C, with the DO being set to 10–20%. Culture samples were taken every 12 h to determine the OD600, dry weight of the cells, and enzyme activity.
Bio-bleaching of pulp
The straw pulp was provided by Wuhan Chenming Paper Co. Ltd. The usage of the xylanase is 15 U/g bone dry pulp. The usage of bleaching solution is 2%., and the enzyme to act on pulp was performed at 50 °C and pH 8.0 for 30 min. The brightness (%ISO), tensile strength and burst factor of paper were measured by YQ-Z-48A ISO Brightness color detector, D-KZW 300 Digital display tensile testing machine, and DCP-SLY1000 tensile testing machine, respectively. All these experiments were performed in triplicates.
The accession number
The GenBank accession number of gene xynHB and xynHBN188As were AY954630 and KU188327, respectively.
Statistical Analysis
All the experiment results were analyzed by MS Excel (MS Office 2010, USA). Differences were considered significant at p < 0.05.
Results
Improvement of thermo-stability by the mutagenesis of N188A
There are three potential glycosylation sites on the XynHB, when expressed in Pichia, it gets glycosylated and presented two target protein bands30. We had intended to study the effects of enzymatic glycosylation on three glycosylation sites in Pichia. When we mutated the third glycosylation site N188 to A, the result showed that the recombinant XynHBN188A in P. pastoris exhibited complete activity after 30 min preincubation at 60 °C, while XynHB in P. pastoris had only 20% residual activity after 30 min preincubation at 60 °C, indicating its enhanced endurance at high temperature. However, the extent of glycosylation of both xylanases were almost similar, consistent with the previous report in which glycosylation sites near C-terminal were not glycosylated or less glycosylated38. So, we predicted the improvement of thermo-stability maybe not related to its glycosylation. In order to determine whether the site mutation caused an increase in thermal stability, we expressed the site mutated protein XynHBN188A in E. coli. Recombinant xylanases were purified and evaluated according to the methods 2.3 (Figure S2). A 26 kDa band was observed, corresponding to the predicted size, 3 kDa larger than the native xylanase (23 kDa), since recombinant xylanase have 34 amino acids more in N-terminal30. The thermo-stability of purified xylanase was evaluated by measuring its residual activities after incubation at 60 °C at different time intervals (Fig. 1). Results indicated that the thermo-stability of the XynHBN188A was 1.5-fold higher than that of XynHB at 60 °C for 30 min. The other characters of XynHBN188A like the optimum temperature (50 °C), pH (8), pH stability, specific activity (2,510 mg/mL) were identified, but no significant difference with the wild-type XynHB.

Thermo-stability of the XynHB and its mutant XynHBN188A.
The thermal stability of XynHB and its mutant XynHBN188A was determined at the 60 °C in 50 mM Tris–HCl buffer (pH 8.0). After incubation, at regular time intervals of 5 min the residual activity of enzyme was measured at pH 8.0 and 50 °C. Each value of the assay was the arithmetic mean of triplicate measurements.
To evaluate the effects of mutation N188A on the improvement of xylanase’s thermo-stability, the structures of XynHB and XynHBN188A were modeled by homology modeling and further analyzed using DS program. Interestingly, the overall three-dimensional structures of XynHB and XynHBN188A are practically identical except the N-terminal region (Fig. 2a). The detailed N-terminus structure of XynHB and XynHBN188A had two major differences as shown in Fig. 2b and c. One difference is that the two hydrogen bonds formed between Arg42 and Ile38, Asp40 were only observed in XynHBN188A, dragging the N-terminus to the middle core structure of the protein. The other is Glu36 formed three new hydrogen with Glu28, Glu29 only in XynHBN188A, strengthening the electrostatic interactions between the amino acids in N-terminal region of the mutant. The mutant expressed in Pichia doesn’t have more 34 amino acids from the vector and its thermo-stability also improved, so we assumed the two hydrogen bonds formed between Arg42 and Ile38, Asp40 might have helped to form an intense structure, responsible for the improved thermo-stability of the mutant XynHBN188A.

Homology modeling and structure comparison of wild type XynHB and XynHBN188A.
(a) The predicted modeling structure of XynHB and XynHBN188A. Dark blue line is the structure of the wild type XynHB while purple is the mutant enzyme XynHBN188A. The mutation sites were shown by ball and line. The 34 amino acid sequence in the N-terminal which were from the vector was marked in yellow. (b) The N-terminus structure of wild type XynHB. (c) The N-terminus structure of mutant XynHBN188A.
Codon optimization enhance the expression of xylanase
In order to further increase the expression level of xylanase in P. pastoris, the gene sequence of xynHBN188A was optimized based on codon bias of P. pastoris. Total 139 nucleotides were substituted in the optimized sequence of xynHBN188As. The G + C content of the xynHBN188As was 39.60% compared to 42.41% in xynHBN188A. The recombinant strains presenting the smallest halo in the RBB-Xylan plate were picked up as described in the Methods 2.6, and the isolated strains containing xynHBN188As and xynHBN188A were named as P81 and F47, respectively. P81 exhibited a xylanase activity of 907 U/ml in the culture supernatant, while F47 had a xylanase activity of 650 U/ml. This result suggested that enzyme activity enhanced by codon optimization was 39.5% (Fig. 3c).

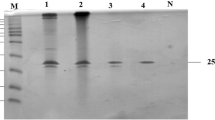
(a) Screening of the high-copy number strain Y16. (b) SDS-PAGE analysis of high-yield strain Y16. M, Protein molecular weight standard. 1, Crude enzyme expressed by P81 strain. 2, Crude enzyme diluted 10 times expressed by Y16 strain. 3, Crude enzyme expressed by Y16. (c) Codon usage and high copy number improves the expression yield of XynHBN188A in P. pastoris.
Screening of recombinant Pichia with high expression of xylanase
The xynHBN188As were integrated into the genome of P. pastoris GS115 and grown on MD plate. Single colonies were re-inoculated on the BMMY plate containing 0.5% RBB-xylan, followed by induction with methanol. The colony forming the largest hydrolysis halo on the plate indicates the highest xylanase activity with more gene copies integrated in the P. pastoris genome, and the identified colony in our study was named as Y16 and set for further evaluation (Fig. 3a).
Further biochemical characterization of high-copy strain Y16 and single-copy strain P81 were performed. The culture supernatants of Y16 and P81 in shaking flask fermentation were detected via SDS-PAGE (Fig. 3b), in which two protein bands with molecular weights of approximate 29.6 kDa and 32.2 kDa were observed. In our previous published study30, we have performed the zymography analysis to confirm that the two protein bands (32.2 kDa and 29.6 kDa) were recombinant xylanase, and EndoH treatment to testify that they were both glycosylated products, and the native xylanase from B. pumilus HBP8 was used as a control. The protein yield of the Y16 strain was around 10 times higher than that of P81 (Fig. 3b). Xylanase activity in the supernatant of Y16 was 6,403 U/ml, while P81 was 907 U/ml (Fig. 3c).
The high copy number of integrated gene in the host chromosome normally led to the higher expression yield of target proteins. Therefore, qPCR was performed to determine the copy number of the exogenous xynHBN188As gene in the genome of Y16. P81 was used as a calibrator strain, and GAP gene of P. pastoris was used as the reference gene. The qPCR analysis was performed according to the Methods 2.8. The mean CT values for the target and internal control genes were calculated and the fold difference was determined as 2ΔΔCT, which was calculated as the equation below, ΔΔCT = (CT,xynHBS − CT,GAP)Test − (CT,xynHBS − CT,GAP)Calibrator28. The results (Table 2) indicated that the copy number of xynHBN188As in the Y16 strain was 9.15.
High-density production of xylanase in Pichia strain Y16
Fed-batch fermentation of the strain Y16 was carried out in a 5 L fermentor by using a three phase growth protocol (Fig. 4). The glycerol batch phase was first employed for 12 h after inoculation. The glycerol fed-batch phase terminated after 35 h when the dry cells weight (DCW) reached 126 g/L. When the glycerol feed was stopped, the DO increased approximately 60%. In the third phase, the methanol fed-batch phase was started to induce the production of xylanase by feeding methanol in a stepwise increasing rate. No apparent xylanase activity was detected until the methanol fed-batch phase was initiated. After the methanol fed-batch phased for 96 h, the cell density reached 159 g/L, and the maximum xylanase activity in the supernatant was 48,241 U/mL (Figs 4 and 5). This enzyme activity was 7.5 times higher than that of the shake flask fermentation, which was only 6,403 U/ml.

DO, OD and xylanase activity curves in a 5 L fermentor.

Xylanase activity and cell dry weight of Y16 in a 5 L fermentor.
Bio-bleaching studies using the straw pulp
The application of XynHBN188A on the straw pulp was also analyzed by adding the fermentation broth of Y16 onto the straw pulp. The results (Table 3) indicated that the paper treated with recombinant xylanase exhibited better quality in brightness, tensile strength and burst factor compared to the control without xylanase addition. The supplement of 15 U/g xylanase enhanced the brightness (%ISO) by 2%, the tensile strength by 13% and the burst factor by 6.5%.
Discussion
The chemicals used for paper bleaching process resulted in the release of large amounts of chlorinated organic compounds that are known to have toxic, mutagenic and carcinogenic effects, causing serious environmental pollution. After being subjected to chemical bleaching, xylanase treated pulp results in reduction of chlorine and chlorine dioxide consumption, along with a reduction in COD value with significant improvement in various pulp properties. Therefore, developing a cost-efficient bleaching process with xylanase represents a positive benefit to paper industry, in which the low-cost alkaline and thermo-stable xylanase is a key issue.
In our earlier study, xylanase was effective in alkaline condition (pH 8.6) with 90% activity and its activity in the supernatant of the recombinant P. pastoris expressing xynHB was 644 U/ml30. However, such thermo-stability and enzyme expression yield did not meet the industrial demand of paper production. The thermo-stability of XynHB was first improved. Previous reports indicated that N-terminal region and hydrogen bond network of xylanase played crucial roles in its thermo-stability8,9,15. GH11 xylanases of thermophilic origin generally have longer N-terminal region compared to mesophilic ones, indicating its role on enhancing the thermo-stability39,40. Several attempts have been made to enhance the thermo-stability of xylanase by substituting the N-terminal region of various non-thermostable xylanases with those of thermostable xylanases8. We first mutated N188 site which led to the increase in the thermo-stability of xylanase XynHB, and its thermo-stability tends to be higher and effective at 60 °C. From the homology modeling analysis between the structures of the wide type and mutant xylanase, there was difference in the N-terminus. The N-terminus of the mutant became more nearer to the middle core structure of the protein, which results in a more intense overall structure. This was similar to the thermo-stability of xylanase from Aspergillus niger and mesophilic xylanase SoxB from S. olivaceovirdis was largely enhanced by substituting their corresponding N-terminal regions with xylanase A (TfxA) from T. fusca7. Furthermore, hydrogen bond network, as one of the most important interactions for maintaining the protein’s secondary structures, can provide significant energetic contribution to the thermo-stability of a protein15,16. Previous studies have indicated that hydrogen bonds were the key to the main differences between B. circulans xylanase and the thermophilic T. lanuginosus xylanase41, the two hydrogen bonds formed between Arg42 and Ile38, Asp40 were only observed in XynHBN188A, dragging the N-terminus to the middle core structure of the protein, which in-turn strengthened the electrostatic interactions between the amino acids in N-terminal region of the mutant. These major structural differences might have helped to form an intense structure, responsible for the improved thermo-stability of the mutant XynHBN188A.
P. pastoris is able to perform many eukaryotic post-translational modifications, including proteolytic processing, disulfide bond formation and glycosylation. Moreover, it can also efficiently secrete heterologous protein with low level of background protein, which makes it for zero need in disrupting the cells, simplifying the purification process of the secreted heterologous protein for paper making. More importantly, P. pastoris is specifically attractive for enzymes used in paper industry because it does not produce endogenous cellulases that weaken the fiber. Thus, P. pastoris is an ideal host for xylanase expression. In the present study, an optimized xynHBN188As was over expressed in P. pastoris by codon optimization and functional screening, with more than nine copies of xylanase gene on the genome of P. pastoris. In the previous studies, the strains containing high gene copies with high protein yield were screened by antibiotics resistance, such as G418 and zeocin. The constructed strains made by this method contained several antibiotic resistance genes, increasing the risk of biological safety. But the constructed strain Y16 generated in this study did not contain any antibiotic resistance gene, avoiding the potential biosafety issues.
Till date, the reported highest xylanase activity was 73,400 U/ml in 3.7 L fermentor, which was obtained by an acidic xylanase, from the acidophilic fungus Bispora sp. MEY-129. The xylanase activity in our recombinant stain reached 48,241 U/mL in 5 L fermentor after 96 h induction, showing the highest xylanase activity at alkaline condition compared to the earlier published reports. The xylanase activity in our recombinant stain reached 48,241 U/mL which is much higher than the xylanase activity in the supernatant of the recombinant P. pastoris expressing xynHB was 644 U/ml from our earlier study before modification30. Structural modification of the protein and the alteration in the fermentation increased the xylanase activity to a larger extent, which could be used in the industrial level as the enzyme being highly stable at pH 7.5 to 8.6 and 50 °C with maximum activity. Further application results in the pulp bleaching process indicated that the recombinant xylanase exhibited better effects in improving the quality of paper.
In summary, due to its alkaline, thermostable properties with high-yield in P. pastoris, the mutant XynHBN188A is a potentially good candidate for industrial application and crude enzyme solution of Y16 strain is good to be used directly in bleaching pulp without purification. In fact, the subsequent pulp bleaching experiments provided direct evidence that crude enzyme solution could improve the qualities of the paper in brightness, tensile strength and burst factor.
Additional Information
How to cite this article: Lu, Y. et al. High-level expression of improved thermo-stable alkaline xylanase variant in Pichia Pastoris through codon optimization, multiple gene insertion and high-density fermentation. Sci. Rep. 6, 37869; doi: 10.1038/srep37869 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Ali, M. & Sreekrishnan, T. Aquatic toxicity from pulp and paper mill effluents: a review. Advances in environmental research 5, 175–196 (2001).
Juturu, V. & Wu, J. C. Microbial xylanases: engineering, production and industrial applications. Biotechnology advances 30, 1219–1227 (2012).
Beg, Q., Kapoor, M., Mahajan, L. & Hoondal, G. Microbial xylanases and their industrial applications: a review. Applied microbiology and biotechnology 56, 326–338 (2001).
Collins, T., Gerday, C. & Feller, G. Xylanases, xylanase families and extremophilic xylanases. FEMS microbiology reviews 29, 3–23 (2005).
Verma, D. & Satyanarayana, T. Molecular approaches for ameliorating microbial xylanases. Bioresource technology 117, 360–367 (2012).
Goswami, G. K. & Pathak, R. R. Microbial xylanases and their biomedical applications: a review. International Journal of Basic & Clinical Pharmacology 2, 237–246 (2013).
Zhang, S. et al. Five mutations in N-terminus confer thermostability on mesophilic xylanase. Biochemical and biophysical research communications 395, 200–206 (2010).
Sun, J.-Y. et al. Improvement of the thermostability and catalytic activity of a mesophilic family 11 xylanase by N-terminus replacement. Protein expression and purification 42, 122–130 (2005).
Gao, S. J. et al. Engineering hyperthermostability into a mesophilic family 11 xylanase from Aspergillus oryzae by in silico design of N‐terminus substitution. Biotechnology and bioengineering 110, 1028–1038 (2013).
Jeong, M.-Y., Kim, S., Yun, C.-W., Choi, Y.-J. & Cho, S.-G. Engineering a de novo internal disulfide bridge to improve the thermal stability of xylanase from Bacillus stearothermophilus No. 236. Journal of biotechnology 127, 300–309 (2007).
Wang, Y. et al. Improved thermal performance of Thermomyces lanuginosus GH11 xylanase by engineering of an N-terminal disulfide bridge. Bioresource technology 112, 275–279 (2012).
Fenel, F., Leisola, M., Jänis, J. & Turunen, O. A de novo designed N-terminal disulphide bridge stabilizes the Trichoderma reesei endo-1, 4-β-xylanase II. Journal of biotechnology 108, 137–143 (2004).
Turunen, O., Vuorio, M., Fenel, F. & Leisola, M. Engineering of multiple arginines into the Ser/Thr surface of Trichoderma reesei endo-1, 4-β-xylanase II increases the thermotolerance and shifts the pH optimum towards alkaline pH. Protein engineering 15, 141–145 (2002).
Wang, K. et al. Thermostability improvement of a streptomyces xylanase by introducing proline and glutamic acid residues. Applied and environmental microbiology 80, 2158–2165 (2014).
Wales, T. E. & Fitzgerald, M. C. The energetic contribution of backbone-backbone hydrogen bonds to the thermodynamic stability of a hyperstable P22 arc repressor mutant. Journal of the American Chemical Society 123, 7709–7710 (2001).
Honig, B. & Yang, A.-S. Free energy balance in protein folding. Advances in protein chemistry 46, 27–58 (1995).
Meng, D. et al. Distinct Roles for Carbohydrate-Binding Modules of GH10 and GH11 Xylanases from Caldicellulosiruptor sp. F32 in Thermostability and Catalytic Efficiency. Applied & Environmental Microbiology 81, 2006–2014 (2015).
Zhang, Z. G., Yi, Z. L., Pei, X. Q. & Wu, Z. L. Improving the thermostability of Geobacillus stearothermophilus xylanase XT6 by directed evolution and site-directed mutagenesis. Bioresource technology 101, 9272–9278 (2010).
Stephens, D. E. et al. Creation of thermostable and alkaline stable xylanase variants by DNA shuffling. Journal of biotechnology 187, 139–146 (2014).
Mchunu, N. P., Singh, S. & Permaul, K. Expression of an alkalo-tolerant fungal xylanase enhanced by directed evolution in Pichia pastoris and Escherichia coli. Journal of biotechnology 141, 26–30 (2009).
Techapun, C., Poosaran, N., Watanabe, M. & Sasaki, K. Thermostable and alkaline-tolerant microbial cellulase-free xylanases produced from agricultural wastes and the properties required for use in pulp bleaching bioprocesses: a review. Process Biochemistry 38, 1327–1340 (2003).
Li, Y., Zhang, B., Chen, X., Chen, Y. & Cao, Y. Improvement of Aspergillus sulphureus endo-β-1, 4-xylanase expression in Pichia pastoris by codon optimization and analysis of the enzymic characterization. Applied biochemistry and biotechnology 160, 1321–1331 (2010).
Gupta, V., Garg, S., Capalash, N., Gupta, N. & Sharma, P. Production of thermo-alkali-stable laccase and xylanase by co-culturing of Bacillus sp. and B. halodurans for biobleaching of kraft pulp and deinking of waste paper. Bioprocess and biosystems engineering 38, 947–956 (2015).
Pribowo, A., Arantes, V. & Saddler, J. N. The adsorption and enzyme activity profiles of specific Trichoderma reesei cellulase/xylanase components when hydrolyzing steam pretreated corn stover. Enzyme and Microbial Technology 50, 195–203 (2012).
Sriyapai, T., Somyoonsap, P., Matsui, K., Kawai, F. & Chansiri, K. Cloning of a thermostable xylanase from Actinomadura sp. S14 and its expression in Escherichia coli and Pichia pastoris. Journal of bioscience and bioengineering 111, 528–536 (2011).
Tian, B. et al. Molecular cloning and overexpression of an endo-β-1, 4-xylanase gene from Aspergillus niger in industrial Saccharomyces cerevisiae YS2 strain. Applied biochemistry and biotechnology 170, 320–328 (2013).
Polizeli, M. et al. Xylanases from fungi: properties and industrial applications. Applied microbiology and biotechnology 67, 577–591 (2005).
Macauley Patrick, S., Fazenda, M. L., McNeil, B. & Harvey, L. M. Heterologous protein production using the Pichia pastoris expression system. Yeast 22, 249–270 (2005).
Luo, H. et al. A thermophilic and acid stable family-10 xylanase from the acidophilic fungus Bispora sp. MEY-1. Extremophiles 13, 849–857 (2009).
Zhang, G. M., Hu, Y., Zhuang, Y. H., Ma, L. X. & Zhang, X. E. Molecular cloning and heterologous expression of an alkaline xylanase from Bacillus pumilus HBP8 in Pichia pastoris. Biocatalysis and Biotransformation 24, 371–379 (2006).
Zhang, G. M., Huang, J., Huang, G. R., Ma, L. X. & Zhang, X. E. Molecular cloning and heterologous expression of a new xylanase gene from Plectosphaerella cucumerina. Applied Microbiology and Biotechnology 74, 339–346 (2007).
Yang, H. et al. High-level expression of Proteinase K from Tritirachium album Limber in Pichia pastoris using multi-copy expression strains. Protein expression and purification 122, 38–44 (2016).
Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. nature 227, 680–685 (1970).
Webb, B. & Sali, A. Comparative protein structure modeling using Modeller. Current protocols in bioinformatics 5.6. 1-5.6. 32 (2014).
Zhang, G., Mao, L., Zhao, Y., Xue, Y. & Ma, Y. Characterization of a thermostable xylanase from an alkaliphilic Bacillus sp. Biotechnology Letters 32, 1915–1920 (2010).
Hoffman, C. S. & Winston, F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformaion of Escherichia coli. Gene 57, 267–272 (1987).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. methods 25, 402–408 (2001).
Daly, R. & Hearn, M. T. W. Expression of heterologous proteins in Pichia pastoris : a useful experimental tool in protein engineering and production. Journal of Molecular Recognition 18, 119–138 (2005).
Blanco, A., Diaz, P., Zueco, J., Parascandola, P. & Pastor, F. J. A multidomain xylanase from a Bacillus sp. with a region homologous to thermostabilizing domains of thermophilic enzymes. Microbiology 145, 2163–2170 (1999).
Liu, L., Zhang, G., Zhang, Z., Wang, S. & Chen, H. Terminal amino acids disturb xylanase thermostability and activity. Journal of Biological Chemistry 286, 44710–44715 (2011).
Vieira, D. S. & Degreve, L. An insight into the thermostability of a pair of xylanases: the role of hydrogen bonds. Molecular Physics 107, 59–69 (2009).
Acknowledgements
This work was supported by the Ministry of Science and Technology of China (863 program 2012AA022203C and 2014AA022203C) and the Key Development Program of Chinese Academy of Sciences (KSZD-EW-Z-015).
Author information
Authors and Affiliations
Contributions
Z.G.M. designed the study, edited the manuscript and was supported by the 863 program 2012AA022203C and 2014AA022203C. L.Y.H. and F.C. conducted the experiments. W.Q.H. and Z.Y.L. wrote the main manuscript text and W.Q.H. was supported by the project KSZD-EW-Z-015. M.Y.H. did the statistical analysis of the data and contributed to design the study. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lu, Y., Fang, C., Wang, Q. et al. High-level expression of improved thermo-stable alkaline xylanase variant in Pichia Pastoris through codon optimization, multiple gene insertion and high-density fermentation. Sci Rep 6, 37869 (2016). https://doi.org/10.1038/srep37869
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37869
This article is cited by
-
Enhanced production of a recombinant xylanase (XT6): optimization of production and purification, and scaled-up batch fermentation in a stirred tank bioreactor
Scientific Reports (2023)
-
Affinity-based and in a single step purification of recombinant horseradish peroxidase A2A isoenzyme produced by Pichia pastoris
Bioprocess and Biosystems Engineering (2023)
-
Constitutive expression of codon optimized Trichoderma reesei TrCel5A in Pichia pastoris using GAP promoter
Systems Microbiology and Biomanufacturing (2022)
-
Co-expressing GroEL–GroES, Ssa1–Sis1 and Bip–PDI chaperones for enhanced intracellular production and partial-wall breaking improved stability of porcine growth hormone
Microbial Cell Factories (2020)
-
N-Terminal Fused Signal Peptide Prompted Extracellular Production of a Bacillus-Derived Alkaline and Thermo Stable Xylanase in E. coli Through Cell Autolysis
Applied Biochemistry and Biotechnology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
