
Abstract
Although manganese oxide- and graphene-based supercapacitors have been widely studied, their charge storage mechanisms are not yet fully investigated. In this work, we have studied the charge storage mechanisms of K-birnassite MnO2 nanosheets and N-doped reduced graphene oxide aerogel (N-rGOae) using an in situ X-ray absorption spectroscopy (XAS) and an electrochemical quart crystal microbalance (EQCM). The oxidation number of Mn at the MnO2 electrode is +3.01 at 0 V vs. SCE for the charging process and gets oxidized to +3.12 at +0.8 V vs. SCE and then reduced back to +3.01 at 0 V vs. SCE for the discharging process. The mass change of solvated ions, inserted to the layers of MnO2 during the charging process is 7.4 μg cm−2. Whilst, the mass change of the solvated ions at the N-rGOae electrode is 8.4 μg cm−2. An asymmetric supercapacitor of MnO2//N-rGOae (CR2016) provides a maximum specific capacitance of ca. 467 F g−1 at 1 A g−1, a maximum specific power of 39 kW kg−1 and a specific energy of 40 Wh kg−1 with a wide working potential of 1.6 V and 93.2% capacity retention after 7,500 cycles. The MnO2//N-rGOae supercapacitor may be practically used in high power and energy applications.
Similar content being viewed by others


Introduction
Supercapacitors or electrochemical capacitors are energy-storage devices widely used in many high-power applications1,2. They have high specific power (~10 kW kg−1) and long cycle life (up to 500,000 cycles)3 when compared with batteries4. This is because the charge storage mechanisms of supercapacitors are mainly at the solid-liquid interface via electrochemical double layer capacitive (EDLC) and pseudocapacitive behaviors. On the other hand, the batteries store charges via redox reactions based on intercalation chemistry5. Improvement in the specific energy of the supercapacitors while keeping their high specific power and capacitance retention is therefore a focal point in the supercapacitor research area.
Supercapacitors are classified to be either symmetric or asymmetric depending on the materials used at the positive and negative electrodes. The difficulty in developing symmetric supercapacitor, which use the identical material at both positive and negative electrodes, is that a single material will only prefer either solvated positive or negative ions. The charge storage performance of the symmetric supercapacitor is therefore limited by the electrode where it can store less charge. To solve this problem, asymmetric supercapacitors (ASCs) using different materials at the positive and negative electrodes are of interest since they can provide higher charge storage performance with wider working potentials6. The maximum charge storage capacity of the ASCs can be finely tuned and achieved by using proper materials and compositions at positive and negative electrodes. Therefore, the recent effort has been devoted to developing the electrode materials of the advanced ASCs. Recently, the ASC of the polypyrrole nanotubes (positive electrode)//N-doped carbon nanotubes (negative electrode) can provide a wide working potential of 1.4 V, a specific energy of 28.95 Wh kg−1 with a specific power of 7.75 kW kg−1 and a cyclic stability of ca. 90% retention after 2,000 cycles7. The ASC of Ni–Co hydroxide@reduced graphene oxide//3D porous carbon exhibits a specific energy of 56.1 Wh kg–1 with 80% retention after 17,000 cycles8. Note, Ni and Co hydroxides are battery-like electrode materials. The ASC of the MnO2 nanosheet//carbon fibers displays a specific capacitance of 87.1 F g−1 and a specific energy of 27.2 Wh kg−1 with 95% capacitance retention over 3,000 cycles9. The ASC of Fe2N //TiN exhibits 5.4 Wh kg−1 and specific power of ca. 6.4 kW kg−1 with 98% capacity retention in 20,000 cycles10. The ASC based on Ti-doped Fe2O3@PEDOT//MnO2 provides an energy density of 0.89 mWh cm−3 with about 85% retention capacitance after 6,000 cycles11. To further improve the charge storage performance of the ASCs, the positive and negative electrode materials with high ionic and electronic conductivities, porosity, and surface area are needed. In this work, new advanced ASCs have been fabricated using MnO2 nanosheets and nitrogen-doped reduced graphene oxide aerogel (N-rGOae) as positive and negative electrodes, respectively.
Among transition metal oxide materials, MnO2 is well recognized as a good candidate for the positive electrode due to its wide potential range in the positive side and high theoretical specific capacitance, high stability, low cost, abundance, and no environmental hazard12,13,14,15. Lee and Goodenough firstly presented that the amorphous MnO2.H2O used as an active material for the supercapacitors in KCl solution exhibited a specific capacitance of ca. 200 F g−1 16. Toupin, M. et al. reported a change in the oxidation state between Mn3+ and Mn4+ during the charge/discharge process of the MnO2 electrode using an ex situ X-ray photoelectron spectroscopy (XPS) measurement of the dried MnO2 electrode after polarized17. They also reported that the charge compensation of the Mn3+ a reduced state is due to Na+ and H+ adsorption17. In contrast, Xu, C. et al. studied the charge storage mechanism of the MnO2 by controlling the pH of the electrolytes and reported that the cations of the electrolyte rather than H+ are responsible for the pseudocapacitance of MnO218. In operando Raman spectroscopy was also employed to probe the structural changes of the α-MnO2 electrode during the charge/discharge process for which the charge storage mechanism is based on the intercalation chemistry19. As the results, it can be concluded here that the charge storage mechanisms of MnO2-based supercapacitors are not yet fully clear.
Interestingly, the mixed valent MnOx including MnO2 and Mn3O4 recently reported exhibits superior charge storage performance than individual MnO2 or Mn3O420,21,22. Among several methods for the preparation of MnOx nanostructures on conductive substrates including precipitation23, sol-gel16, and electrodeposition24, the electrodeposition is well-recognized as an efficiency method with high homogeneity active species. It is also simple, scalable, and cheap technique25. Also, this technique does not require the polymer binders (e.g., PVDF, PTFE), which can introduce many disadvantages including an obstacle for the movement of ions and electronic charge transport. It is also necessary to note here that the charge storage mechanisms of the mixed valent MnOx have not yet been investigated. Thus, understating how the Mn oxidation states do change during the charging/discharging processes is crucial to the development of this material.
For the negative electrodes, the N-rGOae with high surface area and porosity, which are good for supercapacitor electrodes. The diluted N- and O-containing groups of the N-rGOae can lead to high ionic adsorption26. They can also store the electronic charges via surface redox reactions27,28. Interconnected 3D graphene structure can enhance the diffusion of solvated ions via a capillary force providing ultrahigh specific powder2. However, an important question how much solvated ionic charges can be stored by the N-rGOae has not yet been reported. Electrochemical quartz crystal microbalance (EQCM) is then used in this work to address this issue during the charging/discharging processes.
In this work, MnO2 nanosheets with a birnessite structure having negatively charged MnO2 layers along with K+ counter ions and water among the adjacent layers were synthesized by a potential-step electrodeposition. The oxidation number of Mn in MnO2 nanosheets during the charging/discharging processes was subsequently monitored by an in situ X-ray absorption spectroscopy (XAS). In addition, the mass change on the electrodes of MnO2 and N-rGOae during charging/discharging was evaluated by an in situ EQCM. The results provide further understanding on the charge storage mechanisms of MnO2 nanosheets and N-rGOae.
Results and Discussion
Morphologies of as-synthesised materials
The morphology of the MnO2 synthesized using the potential-step electrodeposition was characterized by FE-SEM as shown in Fig. 1a. The as-electrodeposited MnO2 is rather porous with a pore diameter of ca. 10–50 nm due to the interconnection of the MnO2 nanosheets. This morphology is ideal for the supercapacitor electrode since it can enhance the mass transport of the electrolyte due to the capillary force29,30. Figure 1b shows an FE-SEM image of N-rGOae illustrating a few layers of overlapping graphene sheets forming the framework structure with a pore diameter of 0.2–3 μm. The N-rGOae exhibits ultrahigh porosity, which can also accelerate the electrolyte diffusion on the negative electrode. Figure 1c displays a TEM image of the MnO2 for which the morphology of the as-electrodeposited MnO2 is a sheet-like shape with a diameter of ca. 20–50 nm. The MnO2 nanosheets connect to each other forming a porous structure. Figure 1d shows a TEM image of N-rGOae, which is nearly transparent containing many wrinkles of the N-rGOae framework structure. In addition, the EDX mapping of the MnO2 coated on the c-CFP substrate in Fig. 1e displays three main elements, which are C, O, and Mn with 40.2, 27.0, and 18.7% by atomic weight, respectively. The 14.1% remaining element is F, which comes from the carboxyl-modified carbon fiber paper (c-CFP) substrate since a PTFE is used as a binder in the production process of the CFP13.

FE-SEM images of (a) MnO2 and (b) N-rGOae as well as TEM images of (c) MnO2 and (d) N-rGOae and (e) EDX mapping of MnO2/c-CFP mainly containing C, O, and Mn elements.
Structures of as-synthesised materials
To further study the physical and chemical properties of the as-synthesized materials, Raman, XRD, and XPS techniques were carried out. In Fig. 2a, Raman spectra of the MnO2 display two main contribution peaks of the MnO2. N-rGOae displays two distinct peaks at 1,350 and 1,580 cm−1 according to the normal characteristic peaks of the rGO materials. Generally, the D-band at 1,350 cm−1 represents the amount of the disordered carbon structure, which consists of the sp3 carbon atoms at the edge of graphitic sheets. The G-band at 1,580 cm−1 illustrates the vibrational mode of the graphitic sp2carbon sheets31. Additionally, the defect ratio (ID/IG) of N-rGOae is 1.05, which is in good agreement with other previous work32. The amorphous carbon content of N-rGOae calculated from the deconvoluted peaks at around 1,510 cm−1 is ca. 17.7%.

(a) Raman spectra, (b) XRD patterns of MnO2 and N-rGOae and (c) N2 sorption isoterm and pore size distribution of N-rGOae.
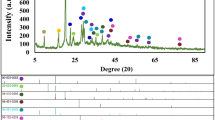
The XRD pattern of N-rGOae (Fig. 2b) displays two broad peaks at 2θ of 24.3 and 43.7° referring to the characteristics of rGO33. The XRD pattern of the as-electrodeposited MnO2 indicates the K-birnessite MnO2 nanosheets (JCPDS 80-1098)34,35. The peaks at 2θ about 12, 24, 37, 43, 56, and 66° are due to (0 0 1), (002), (111), (−112), (113), (020), and (220) planes showing a lamellar structure. The structure consists of single sheets of edge-sharing [MnO6] octahedral and water molecules and K+ between the adjacent layers34,35,36. The orthogonal distance between two consecutive slabs of [MnO6] is ca. 7.3 Å. The mixed vacancy of manganese ions in K-birnassite MnO2 nanosheets plays an important role of a spontaneous redox reaction enhancing the pseudocapacitance of ASCs22. In addition, the N2 gas adsorption was carried out to determine the specific surface area and pore size distribution of N-rGOae as shown in Fig. 2c and d. By following the IUPAC classification, the gas adsorption isotherm of N-rGOae is in a type-IV isotherm (a hysteresis loop type II) owing to interconnected pore networks (see Fig. 2c). The BET surface area of N-rGOae is about 352 m2 g−1 having an average pore width of 3.7 nm (Fig. 2d).
Surface analysis of as-fabricated electrodes
The surface chemical composition of the as-fabricated electrodes was analyzed by the XPS technique. The C1s spectra of N-rGOae sprayed on c-CFP (Fig. 3a) display four main peaks at 284.9, 285.2, 286.1, and 288.5 eV corresponding to C-C, C-N, C-O and C=O, respectively37. The diluted nitrogen content in Fig. 3b can be deconvoluted into four peaks of pyridinic N (399.8 eV), pyrolic N (400.5 eV), graphitic N (401.7 eV) and oxidized N (405.4 eV), respectively38. The graphitic N can improve the charge transfer in the rGO matrix39. Other N-functional groups (Pyrolic N and Pyridinic N) also play an important role for the pseudocapacitance27,28,40.

(a) C1s and (b) N1s XPS spectra of N-rGOae on c-CFP, (c) XPS survey, and (d) Mn2p XPS of MnO2.
A wide-scan XPS spectrum of MnO2 coated on c-CFP is shown in Fig. 3c confirming the elements of the as-prepared electrode. Notably, K is also found on the XPS spectrum since K+ is a balance charge of negatively-charged MnO2 layers. The Mn2p spectra of the as-electrodeposited MnO2 coated on c-CFP in Fig. 3d show two broad peaks of Mn2p3/2 and Mn2p1/2, which can be deconvoluted to many peaks corresponding to two different oxidation states of Mn species41. This material has the advantage of mixed valent MnO2 for supercapacitors. Each broad peak can be classified into three parts at 640.9, 642.1, and 645.9 eV for Mn2p3/2 and 652.6, 653.8, and 656.2 eV for Mn2p1/2. The peaks at 640.9 and 652.6 eV are the characteristics of Mn3+ (21.3%) while those at 642.1 and 653.8 eV are the characteristics of Mn4+ (78.7%) as well as those at 645.9 and 656.2 eV are attributed to shakeup satellites. For the O1s XPS spectrum, which is not shown here, the spectrum contains three main peaks located at 531.4, 532.6, and 533.1 eV attributing to Mn-O-H, H-O-H and O-C, respectively42.
Electrochemical evaluatioon of as-fabricated electrodes
To evaluate the electrochemical property of the as-fabricated electrodes, a three-electrode system using the as-synthesized material as a working electrode, a Pt wire as a counter electrode, and a saturated calomel electrode (SCE) as a reference electrode was carried out in 0.5 M Na2SO4 solution. The optimized potential range of the as-prepared electrodes is shown in CVs (Fig. 4a). Also, an optimum mass ratio finely tuned between the mass of active materials at positive and negative electrodes (m+/m−) is 1.75 providing the highest charge storage performance calculated by a charge balance according to Eq. 143 as follow;

(a) CVs of the as-fabricated electrodes at 25 mV s−1 and (b) CVs at different working potentials (50 mV s−1), (c) GCDs at different working potentials (5 A g−1), and (d) specific capacitance and coulombic efficiency vs. applied current density, (e) the b value as a function of potential, and (f) the bar chart of the diffusion-controlled intercalation capacitance vs. scan rates of as-fabricated MnO2//N-rGOae supercapacitor devices.

where m is the mass of the active material, Cs is the specific capacitance, and ΔV is the voltage range for positive and negative electrodes44. Note, all electrochemical properties of half-cell MnO2 and N-rGOae electrodes are shown in Figure S1 and 2 of the supporting information, respectively. The MnO2//N-rGOae supercapacitor was then assembled using a hydrolyzed PE containing 0.5 M Na2SO4 as a separator. The operating potential window was varied from 0.8–1.6 V as shown in Fig. 4b. The rectangular-shaped CVs with a broad redox peak indicate the pseudocapacitive behavior that comes from the surface redox reaction of MnO215. Surprisingly, the as-fabricated device levels off at the same range of the current density until 1.6 V. Moreover, GCDs show a symmetrical shape relating to the CV result (Fig. 4c). The MnO2//N-rGOae device exhibits the specific capacitances of 467.38–342.43 F g−1 (4.55–3.33F cm−3) at 1.0–5.0A g−1, respectively. The device has high coulombic efficiency up to 100% at 5 A g−1 (Fig. 4d). To further study the capacitive effect, b value was calculated from a power law according to Eq. 245 as follow;

where i is the current and ν is the scan rate. Both a and b are the constant parameters for which the b value can be determined from a slope of the linear plot between log i vs. log ν (see inset graphs in Fig. 4e and Figure S3a). According to power law relationship, i is equal to aν for non-diffusion limited processes and i is equal to aν1/2 for diffusion limited processes. Typically, the b value is equal to 1.0 for non-diffusion-controlled surface capacitive and equal to 0.5 for diffusion-controlled redox reaction, which is a typical battery behavior44. The result in Fig. 4e shows that the b values in this work are 0.56, 0.89, 0.95, 0.95, 0.84, 0.80 at the potentials of 0.25, 0.50, 0.75, 1.00, 1.25, and 1.50 V, respectively. This can confirm that the devices have both EDLC and surface redox reactions. In addition, the percentage of intercalation and capacitive contribution calculated by Eq. 346 is shown in Fig. 4f. The intercalation capacitance decreases when increasing scan rates due to the diffusion limit of the electrolytes.

where k1 and k2 are the slope and interception, respectively, which can be determined from Figure S3b.
Besides, the EIS result of the MnO2//N-rGOae supercapacitor at a sinusoidal signal of 10 mV from 100 kHz to 1 mHz is shown in the Nyquist plots (Fig. 5a). The straight line of the Nyquist plots increases sharply at a low frequency region to the Y-axis indicating almost the ideal supercapacitor dominated by the capacitive behavior from the formation of ionic and electronic charges. At high frequency, the electronic charge transfer resistance (Rct) due to the surface redox of MnO2 is about 13.45 Ω with an internal resistance (Rs) of 2.32 Ω located at the interception on the X-axis. In addition, the relaxation-time constant (τ0), which is a minimum time required to discharge for all stored charges, can be determined from an inversion of the frequency at the maximum phase angle as shown in Fig. 5b. It is necessary to note the smaller value of τ0 the higher power of the supercapacitors47. In this work, τ0 is about 686 ms, which is much smaller than other previous report1,48. Finally, the stability of the MnO2//N-rGOae supercapacitor evaluated by the GCD method over 7,500 cycles at 5 A g−1 (Fig. 5c) is over 93.2% retention. The as-assembled device provides the highest specific energy of 40 Wh kg−1 and the highest specific power of 39 kW kg−1, which are much higher than those of other previous related report (see Fig. 5d)49,50,51. Note, CVs at different scan rates, GVDs at different specific currents, and the calculated specific capacitances at different frequencies of the device are also shown in Figure S4 of the supporting information.

In situ X-ray absorption spectroscopy
In order to clarify the origin of the remarkable specific energy and specific power of the MnO2-based supercapacitor, the charge storage behavior occurred during the charge/discharge processes has been investigated by the in-situ XAS measurement. As Mn in the manganese oxide with different oxidation states plays a prominent role for the surface redox reaction during the charge/discharge process, in situ monitoring the oxidation number change of the Mn during charge/discharge processes is therefore crucial for understanding the pseudocapacitive behavior. In this work, the in situ XAS technique was carried out together with the chronoamperometry in 0.5 M Na2SO4 electrolyte during applying the potentials stepped from 0.0, 0.4, and 0.8 V vs. SCE and the backward potentials from 0.8 to 0.4 V vs. SCE and from 0.4 to 0.0 V vs. SCE. Note, in order to reach the steady state, each step potential was hold for 15 min before starting the XAS measurement52,53. The Mn K-edge fluorescence energy of the MnO2 charged at 0.0 V vs. SCE is 6548.05 eV and the energy value increases up to 6548.36 and 6548.53 eV when the potentials were applied to 0.4 and 0.8 V vs. SCE, respectively (see Fig. 6a). The Mn oxidation states of the MnO2 electrode being charged at 0.0, 0.4, and 0.8 V vs. SCE are +3.01, +3.08, and +3.12, respectively (see Fig. 6b). Note, the oxidation number of the as-electrodeposited MnO2 is +3.79.

(a) In situ high-resolution Mn K-edge fluorescence XAS spectra of the as-prepared MnO2 electrodes and Mn standard compounds and (b) the oxidation states vs. ΔE (eV) of the MnO2 electrodes during charging/discharging by a chronoamperometry method at applied potentials from 0.0–0.8 V vs. SCE and backward potentials. Note, the XAS was carried out after reaching the steady state.
The XAS result here confirms the reversible redox reaction of the MnO2 and the proposed general redox reaction (4) below based on the intercalation/de-intercalation processes of Na+ and H+ is shown below15,16,54,55;

When the stepped potentials were applied backward from 0.8 to 0.4 V vs. SCE and afterward from 0.4 to 0.0 V vs. SCE, the edge energies are 6548.21 eV and 6548.05 eV, respectively (see Fig. 6a). The oxidation states of Mn in the MnO2 return to +3.04 at 0.4 V vs. SCE and +3.01 at 0 V vs. SCE (see Fig. 6b).
In order to clarify the charge storage mechanism of the as-prepared K-birnessite MnO2, the effect of the pH of 0.5 M Na2SO4 electrolyte was also studied by verying pH of the 0.5 M Na2SO4 electrolytes by adding conc. H2SO4 (see the experimental results in Figure S5 and 6 of the supporting information). It is found that at pH beween 0.08 and 1.10, the solvated H+ plays an important role in the charge storage capacity via an intercalation redox reaction (see redox peaks in CVs of Figure S5a) according to the reaction mechanism (5) below;

At pH 2.03–4.02, the solvated Na+ plays a major role to the charge storage capacity of the MnO2 (see the mechanism in reaction (4) above). At pH > 5.36 adjusted by adding NaOH, it was found that the specific capacitances are significantly reduced since the MnO2 layers having negative charge do not like to adsorb/absorb solvated anions i.e., OH−. As the results, we can conclude that H+ plays a significant role in the charge storage capacity at pH < 2.03.
Electrochemical quartz crystal microbalance
In addition to the in situ XAS results, the in situ gravimetric measurement of the mass changes on N-rGOae and MnO2 electrodes was eventually evaluated via the EQCM method. The EQCM electrode was prepared by a drop-coating of the as-prepared materials onto the Au/TiO2 quartz crystal surfaces. The in situ probing via the CV method was carried out in 0.5 M Na2SO4 solution using a three-electrode system with Ag/AgCl (3 M KCl) as the reference electrode and Au wire as the counter electrode (Fig. 7). The quartz resonance frequency (Δf) can be converted into the mass change (Δm) according to the derived Sauerbrey equation (6)56 below;

CVs at 25 mVs−1 and in situ EQCM responses of (a) N-rGOae and (b) MnO2 electrodes.

where the frequency ( ) in Hz and the calibration constant (Cf) is 0.0815 Hz ng−1cm2.
) in Hz and the calibration constant (Cf) is 0.0815 Hz ng−1cm2.
The CV and Δm from the quartz frequency response of N-rGOae are shown in Fig. 7a from −0.1 V to −0.5 V vs. Ag/AgCl. The CV shows a narrow potential window (about 0.6 V) because of small amount of N-rGOae coated onto the Au/TiO2 electrode57. For the charge storage mechanisms of the N-rGOae at the negative electrode, it can store ionic charges via the physical adsorption (EDLC) at the solid-liquid interface by adsorbing/absorbing the solvated ions58. Furthermore, the N-containing groups of the N-doped rGO can store electronic charges via the redox reaction (7) below58;

The Δm or mass deposited to the electrode during the charge process gradually increases to 8.4 μg cm−2. After discharged, the ion accumulation releases to the electrolyte and the electrode returns to the initial state. Besides, the CV of the MnO2 electrode in Fig. 7b displays an anodic potential range from −0.1 to 0.5 V vs. Ag/AgCl. The Δm is 7.4 μg cm−2, relating to solvated cations (i.e. Na+ and K+) inserted/released from the MnO2 layers. This is why the Mn oxidation state of the MnO2 electrode being charged is increased. After fully discharged, the Δm returns to the initial value again confirming the XAS result.
Conclusions
High-performance asymmetric supercapacitor of the MnO2//N-rGOae has been successfully fabricated. The MnO2 nanosheets were prepared using a potential step electrodeposition and used as the positive electrode of the supercapacitor. The N-rGOae was synthesized using a hydrothermal process by reducing graphene oxide with hydrazine (a nitrogen source) and used as the negative electrode. The in situ XAS carried out together with the chronoamperometry indicates that the oxidation state of manganese ions in the MnO2 electrode being charged remarkably rises from +3.01 to +3.12 when applying potentials at 0 to 0.8 V vs. SCE and returns to +3.01 at 0 V vs. SCE during the discharge process. This is a reason why MnO2 nanosheets exhibit excellent capacity retention. The mass changes of solvated ions at the N-rGOae- and MnO2-coated Au/TiO2 quartz crystal EQCM electrodes during the charge/discharge processes are ca. 8.4 and 7.4 μg cm−2, respectively. It is also found in this work that [H+] plays a significant role in the charge storage capacity at pH of the electrolyte, 0.5 M Na2SO4(aq) <2.03. At pH 2.03–4.02, the solvated Na+ plays a major role to the charge storage capacity of the MnO2. At pH > 5.36, the specific capacitance of the device is significantly reduced since the birnassite MnO2 layers having negative charge do not like to adsorb/absorb solvated anions i.e., OH−. An as-fabricated MnO2//N-rGOae with a finely tuned mass loading ratio of 1.75 provides a wide working potential of 1.6 V with the highest specific power and energy of 39 kW kg−1 and 40 Wh kg−1, respectively. This device with a CR2016 size has 93.2% capacity retention after 7,500 cycles at 5 A g−1. The enhancement in the specific energy and specific power of the MnO2//N-rGOae supercapacitors can compete with the batteries in many applications.
Methods
Preparation of flexible carboxyl-modified carbon fiber paper (c-CFP)
The c-CFP substrate was prepared by an acid treatement13,29. Briefly, conc. H2SO4 (150 ml) and conc. HNO3 (50 ml) were mixed together in a beaker by stirring at 100 rpm for 10 min. The CFP with 5 × 5 cm2 was then immersed to the acid mixture and kept stirring at 60 °C at 100 rpm for 1 h. The c-CFP was then washed with Milli-Q water 5 times and dried at 50 °C for 24 h.
Preparation of N-rGOae negative electrode
GO was firstly synthesized using a modified Hummers method previously reported by our group14,26,59,60,61,62. The N-rGOae was then synthesized via a hydrothermal reduction of GO with 0.5 M hydrazine (N2H4) a reducing agent. First, the as-synthesized GO (160 mg) was dispersed in Milli-Q water (80 ml) using a sonication process (100 w) for 2 h. N2H4 was then added to the mixture at room temperature. The mixture was consequently transferred to a Teflon autoclave (100 ml) and heated at 110 °C for 24 h to form N-rGO hydrogel. For the purification, the as-synthesized hydrogel was immersed in Milli-Q water to remove the residuals for 72 h. Finally, the hydrogel was frozen at 0 °C for 24 h. Then, the frozen hydrogel was put in a freezing dryer to remove water at −55 °C for 48 h. The product is so-called N-rGOae. In order to fabricate the negative electrode, the as-synthesized N-rGOae (3 mg) was dispersed in ethanol (3 ml), spray-coated on the c-CFP using an airbrush with a 0.3-mm brush nozzle (Paasche Airbrush Company, USA) and eventually dried at 50 °C for 24 h.
Potential-step electrodeposition of the MnO2 positive electrode
The c-CFP at a diameter of 1.58 cm was immersed in an electrodeposition solution, 30 ml of 250 mM Mn(NO3)2·H2O in 250 mM KCl. MnO2 nanosheets were electrodeposied on the c-CFP by a potential-step electrodeposition at 1.0 V vs. SCE for 3 min and then suddenly switched to 0.5 V vs. SCE for 1 min for which a chronoamporometry method was carried out using a potentiostat (PGSTAT 302 N). In order to have 1–2 mg of MnO2, this process was repeated for 10 times. Finally, the as-electrodeposited electrode was then washed 3 times with Milli-Q water to remove the residual KCl and dried at 50 °C for 24 h.
Morphological and structural characterizations
X-ray diffraction (XRD) using a D8 ADVANCE with DAVINCI design (Bruker optics, Germany) with CuKα of 1.5418 Å was used to characterize the crystalline structures of the as-synthesised materials i.e., GO, N-rGOae, and MnO2. The data were collected from 5 to 80° (2θ) with 0.01 increment. Note, the Si wafer was used as a holder for XRD measurement. Raman spectroscopy was also carried out using a laser excitation wavelength of 532 nm (Senterra Dispersive Raman, Bruker optics, Germany). The field-emission scanning electron microscopy (FE-SEM) images of the as-prepared materials were performed with an accelerating voltage of 15.0 kV (JSM-7001F, JEOL Ltd., Japan). The samples were mounted on the clean surface of carbon conductive tab and placing on the SEM pin stub. Note that the specimens were coated with the platinum by a sputtering technique for 40 sec in order to remove the charging effect. The transmission electron microscopy (TEM) images of the samples were performed with an accelerating voltage of 100 kV (a JEM 1220, JEOL Ltd., Japan). The TEM specimens were prepared by dropping the suspension (∼0.05 mg/ml) of N-rGOae, and MnO2 in ethanol in the copper grids and dried at 50 °C for 3 h. The functional groups and elemental compositions of the as-synthesised materials were also analyzed by X-ray photoelectron spectroscopy (XPS) using an AXIS Ultra DLD (Kratos Analytical Ltd., Manchester, UK) with Al-K alpha radiation (hv = 14,866 eV). In addition, in situ Mn K-edge fluorescent x-ray photoelectron spectroscopy (XAS) measurement was performed at a beamline No. 5 at the Synchrotron Light Research Institute (Public Organization), Nakhon Ratchasima, Thailand using a Ge(220) double-crystal monochromator (energy range 3440–12100 eV). The spectroscopic data were collected in fluorescence mode with a 4-element silicon drift detector. The 4-element silicon drift detector was placed 90° to the beam and 45° to the sample. The Mn K-edge (6539 eV) was calibrated using the Mn foil before measurement. The light dimension on the sample was adjusted to 5 mm width and 1 mm height. The advantage of using in situ XAS measurements is that it can probe or localize the Mn element of the MnO2 electrode during charging/discharging.
For the in situ electrochemical XAS measurement, a chronoamperometry method was used at different potentials (i.e., 0, 0.4, and 0.8 V vs. SCE) to evaluate the electrochemical property of the electrodes. In this measurement, a 3-electrode system using SCE as a reference electrode, Pt wire as a counter electrode, and the as-prepared MnO2 working electrode was carried out in a 0.5 M Na2SO4 (aq.) electrolyte. Note, the electrochemical cell was made from acrylic sheets with the dimension of 2 × 2 × 3.5 cm3 having a drilled hole diameter of 0.8 cm on one 2-cm2 side of the acrylic sheet. The drilled hole was covered by a larger piece of Kapton tape with a diameter of 1.2 cm. The SCE and Pt wire were placed beside the MnO2 electrode at a distance of ca.1 cm but away from the path of the X-rays. In order to get a steady-state current, the MnO2 working electrode was kept at a given potential of interest for at least 15 min before the in situ XAS and chronoamperometry measurements.
Fabrication of ASCs and the electrochemical evaluation
The ASCs were assembled of the negative and positive electrodes with a coin-cell size (CR2016). Hydrolyzed polyethylene (PE) film with a thickness of 25 μm was used as the separator of aqueous-based supercapacitors and 0.5 M Na2SO4(aq) was used as the electrolyte. The electrolyte seperator was prepared by soaking the hydrolized PE in 0.5 M Na2SO4(aq) for 10 min before assembled. Then, the separator was inserted betweent positive and negative electrode. Finally, the coin cell was then assembled by pressing with crimper machine at 100 psi. The electrochemical evaluation of the as-fabricated supercapacitors was carried out using a Metrohm AUTOLAB potentiostat (PGSTAT 302 N) made in Netherlands running NOVA software (version 1.11). Cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) were performed.
Calculation of supercapacitor performances
The specific capacitance (Ccv) of the supercapacitor cell excluding the influence of the c-CFP substrate can be determined from the CV by following calculation Eq. (8)40,63,64,65.

where ΔV is the working potential determined from the discharge potential chosen in the potential range without H2 and O2 evolution, IdV is an area under the discharging curve,  is a scan rate (V/s), and m is a total active mass at negative and positive electrodes (g).
is a scan rate (V/s), and m is a total active mass at negative and positive electrodes (g).
The specific capacitance can also be calculated from the GCD method (CGCD) by following Eq. (9)40,65,66;

where I is the applied constant current (A), Δt is the discharging time (s), and ΔVcell is the potential window (V) excluding iR drop. Note, the iR drop increases when increasing the applied current rate.
The specific capacitance determined from the EIS technique (CEIS) can be calculated from Eq. (10)2,67,68;

where f is the applied frequency and Z″ is the imaginary component of the impedance at the frequency f, which is a negative value. In addition, the equivalent series resistance (ESR) of the supercapacitors was simply determined from the intercept at the X-axis of the Nyquist plots.
Besides, the specific energy (E) of the supercapacitors were calculated by following Eq. (11)69,70,71;

The maximum power (Pmax) at the discharge efficiency of 50% from a maximum voltage at the fully charged state can be calculated as the following Eq. (12)69 ;

where Vmax is a maximum voltage of the cell, and Rcell is a resistance of the cell, which can be determined from the iRcell drop observed in the GCD13.
Additional Information
How to cite this article: Iamprasertkun, P. et al. Charge storage mechanisms of manganese oxide nanosheets and N-doped reduced graphene oxide aerogel for high-performance asymmetric supercapacitors. Sci. Rep. 6, 37560; doi: 10.1038/srep37560 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Pech, D. et al. Ultrahigh-power micrometre-sized supercapacitors based on onion-like carbon. Nat. Nanotechnol. 5, 651–654 (2010).
Wang, X. et al. Three-dimensional strutted graphene grown by substrate-free sugar blowing for high-power-density supercapacitors. Nat. Commun. 4, doi: 10.1038/ncomms3905 (2013).
Pandolfo, A. G. & Hollenkamp, A. F. Carbon properties and their role in supercapacitors. J. Power Sources 157, 11–27 (2006).
Liu, C., Yu, Z., Neff, D., Zhamu, A. & Jang, B. Z. Graphene-Based Supercapacitor with an Ultrahigh Energy Density. Nano Lett. 10, 4863–4868 (2010).
Kotz, R. & Carlen, M. Principles and applications of electrochemical capacitors. Electrochim. Acta 45, 2483–2498 (2000).
Fan, Z. et al. Asymmetric Supercapacitors Based on Graphene/MnO2 and Activated Carbon Nanofiber Electrodes with High Power and Energy Density. Adv. Funct. Mater. 21, 2366–2375 (2011).
Dubal, D. P. et al. Synthetic approach from polypyrrole nanotubes to nitrogen doped pyrolyzed carbon nanotubes for asymmetric supercapacitors. J. Power Sources 308, 158–165 (2016).
Ma, H. et al. Nickel Cobalt Hydroxide @Reduced Graphene Oxide Hybrid Nanolayers for High Performance Asymmetric Supercapacitors with Remarkable Cycling Stability. Acs App. Mater. Interfaces 8, 1992–2000 (2016).
Yu, N. et al. High-Performance Fiber-Shaped All-Solid-State Asymmetric Supercapacitors Based on Ultrathin MnO2 Nanosheet/Carbon Fiber Cathodes for Wearable Electronics. Adv.Energy Mater. 6, doi: 10.1002/aenm.201501458 (2016).
Zhu, C. et al. All Metal Nitrides Solid-State Asymmetric Supercapacitors. Adv. Mater. 27, 4566–4571 (2015).
Zeng, Y. et al. Advanced Ti-Doped Fe2O3@PEDOT Core/Shell Anode for High-Energy Asymmetric Supercapacitors. Adv.Energy Mater. 5, doi: 10.1002/aenm.201402176 (2015).
Phattharasupakun, N. et al. Turning conductive carbon nanospheres into nanosheets for high-performance supercapacitors of MnO2 nanorods. Chem. Commun. 52, 2585–2588 (2016).
Suktha, P. et al. High-Performance Supercapacitor of Functionalized Carbon Fiber Paper with High Surface Ionic and Bulk Electronic Conductivity: Effect of Organic Functional Groups. Electrochim. Acta 176, 504–513 (2015).
Sawangphruk, M. et al. High-performance supercapacitor of manganese oxide/reduced graphene oxide nanocomposite coated on flexible carbon fiber paper. Carbon 60, 109–116 (2013).
Lang, X., Hirata, A., Fujita, T. & Chen, M. Nanoporous metal/oxide hybrid electrodes for electrochemical supercapacitors. Nat. Nanotechnol. 6, 232–236 (2011).
Lee, H. Y. & Goodenough, J. B. Supercapacitor Behavior with KCl Electrolyte. J. Solid State Chem. 144, 220–223 (1999).
Toupin, M., Brousse, T. & Bélanger, D. Charge Storage Mechanism of MnO2 Electrode Used in Aqueous Electrochemical Capacitor. Chem. Mater. 16, 3184–3190 (2004).
Xu, C., Wei, C., Li, B., Kang, F. & Guan, Z. Charge storage mechanism of manganese dioxide for capacitor application: Effect of the mild electrolytes containing alkaline and alkaline-earth metal cations. J. Power Sources 196, 7854–7859 (2011).
Chen, D. et al. Probing the Charge Storage Mechanism of a Pseudocapacitive MnO2 Electrode Using in Operando Raman Spectroscopy. Chem. Mater. 27, 6608–6619 (2015).
Yang, J. et al. All-Solid-State High-Energy Asymmetric Supercapacitors Enabled by Three-Dimensional Mixed-Valent MnOx Nanospike and Graphene Electrodes. ACS App. Mater. Interfaces 7, 22172–22180 (2015).
Hu, C.-C., Hung, C.-Y., Chang, K.-H. & Yang, Y.-L. A hierarchical nanostructure consisting of amorphous MnO2, Mn3O4 nanocrystallites, and single-crystalline MnOOH nanowires for supercapacitors. J. Power Sources 196, 847–850 (2011).
Hu, Y. & Wang, J. MnOx nanosheets for improved electrochemical performances through bilayer nano-architecting. J. Power Sources 286, 394–399 (2015).
Li, Y. et al. Manganese oxide nanowires wrapped with nitrogen doped carbon layers for high performance supercapacitors. J. Colloid Interface Sci. 455, 188–193 (2015).
Ali, G. A. M., Yusoff, M. M., Ng, Y. H., Lim, H. N. & Chong, K. F. Potentiostatic and galvanostatic electrodeposition of manganese oxide for supercapacitor application: A comparison study. Cur. App. Phys. 15, 1143–1147 (2015).
Qin, Y.-H. et al. Electrophoretic deposition of network-like carbon nanofibers as a palladium catalyst support for ethanol oxidation in alkaline media. Carbon 48, 3323–3329 (2010).
Iamprasertkun, P., Krittayavathananon, A. & Sawangphruk, M. N-doped reduced graphene oxide aerogel coated on carboxyl-modified carbon fiber paper for high-performance ionic-liquid supercapacitors. Carbon 102, 455–461 (2016).
You, B., Wang, L., Yao, L. & Yang, J. Three dimensional N-doped graphene-CNT networks for supercapacitor. Chem. Commun. 49, 5016–5018 (2013).
Lee, Y.-H., Chang, K.-H. & Hu, C.-C. Differentiate the pseudocapacitance and double-layer capacitance contributions for nitrogen-doped reduced graphene oxide in acidic and alkaline electrolytes. J. Power Sources 227, 300–308 (2013).
Kaewsongpol, T. et al. High-performance supercapacitor of electrodeposited porous 3D polyaniline nanorods on functionalized carbon fiber paper: Effects of hydrophobic and hydrophilic surfaces of conductive carbon paper substrates. Mater. Today Commun. 4, 176–185 (2015).
Srimuk, P., Luanwuthi, S., Krittayavathananon, A. & Sawangphruk, M. Solid-type supercapacitor of reduced graphene oxide-metal organic framework composite coated on carbon fiber paper. Electrochim. Acta 157, 69–77 (2015).
Yin, F. et al. Self-assembly of mildly reduced graphene oxide monolayer for enhanced Raman scattering. J. Solid State Chem. 237, 57–63 (2016).
Ji, C.-C. et al. Self-assembly of three-dimensional interconnected graphene-based aerogels and its application in supercapacitors. J. Colloid Interface Sci. 407, 416–424 (2013).
Sui, Z.-Y. et al. Nitrogen-Doped Graphene Aerogels as Efficient Supercapacitor Electrodes and Gas Adsorbents. ACS App. Mater. Interfaces 7, 1431–1438 (2015).
Zhang, X. et al. Rapid hydrothermal synthesis of hierarchical nanostructures assembled from ultrathin birnessite-type MnO2 nanosheets for supercapacitor applications. Electrochim. Acta 89, 523–529 (2013).
Dubal, D. P. et al. 3D hierarchical assembly of ultrathin MnO2 nanoflakes on silicon nanowires for high performance micro-supercapacitors in Li- doped ionic liquid. Sci. Rep. 5, 9771 (2015).
Julien, C. et al. Raman spectra of birnessite manganese dioxides. Solid State Ion. 159, 345–356 (2003).
Some, S., Kim, Y., Hwang, E., Yoo, H. & Lee, H. Binol salt as a completely removable graphene surfactant. Chem. Commun. 48, 7732–7734 (2012).
Liu, Y., He, D., Wu, H., Duan, J. & Zhang, Y. Hydrothermal Self-assembly of Manganese Dioxide/Manganese Carbonate/Reduced Graphene Oxide Aerogel for Asymmetric Supercapacitors. Electrochim. Acta 164, 154–162 (2015).
Fan, W., Miao, Y.-E., Huang, Y., Tjiu, W. W. & Liu, T. Flexible free-standing 3D porous N-doped graphene-carbon nanotube hybrid paper for high-performance supercapacitors. RSC Adv. 5, 9228–9236 (2015).
Béguin, F., Presser, V., Balducci, A. & Frackowiak, E. Carbons and Electrolytes for Advanced Supercapacitors. Adv. Mater. 26, 2219–2251 (2014).
Liang, M., Liu, X., Li, W. & Wang, Q. A Tough Nanocomposite Aerogel of Manganese Oxide and Polyaniline as an Electrode for a Supercapacitor. ChemPlusChem 81, 40–43 (2016).
Wang, H.-Q. et al. Direct growth of flower-like 3D MnO2 ultrathin nanosheets on carbon paper as efficient cathode catalyst for rechargeable Li-O2 batteries. RSC Adv. 5, 72495–72499 (2015).
Chen, H. et al. In situ growth of NiCo2S4 nanotube arrays on Ni foam for supercapacitors: Maximizing utilization efficiency at high mass loading to achieve ultrahigh areal pseudocapacitance. J. Power Sources 254, 249–257 (2014).
Wang, R., Xu, C. & Lee, J.-M. High performance asymmetric supercapacitors: New NiOOH nanosheet/graphene hydrogels and pure graphene hydrogels. Nano Energy 19, 210–221 (2016).
Sathiya, M., Prakash, A. S., Ramesha, K., Tarascon, J. M. & Shukla, A. K. V2O5-Anchored Carbon Nanotubes for Enhanced Electrochemical Energy Storage. J. Am. Chem. Soc. 133, 16291–16299 (2011).
Augustyn, V., Simon, P. & Dunn, B. Pseudocapacitive oxide materials for high-rate electrochemical energy storage. Energy Environ. Sci. 7, 1597–1614 (2014).
Lee, G. et al. High-performance all-solid-state flexible micro-supercapacitor arrays with layer-by-layer assembled MWNT/MnOx nanocomposite electrodes. Nanoscale 6, 9655–9664 (2014).
Yuan, L. et al. Flexible Solid-State Supercapacitors Based on Carbon Nanoparticles/MnO2 Nanorods Hybrid Structure. ACS Nano 6, 656–661 (2012).
Yu, Z. et al. Functionalized graphene aerogel composites for high-performance asymmetric supercapacitors. Nano Energy 11, 611–620 (2015).
Yang, X. et al. High-performance aqueous asymmetric supercapacitor based on spinel LiMn2O4 and nitrogen-doped graphene/porous carbon composite. Electrochim. Acta 180, 287–294 (2015).
Kalubarme, R. S., Jadhav, H. S. & Park, C.-J. Electrochemical characteristics of two-dimensional nano-structured MnO2 for symmetric supercapacitor. Electrochim. Acta 87, 457–465 (2013).
Wongsaprom, K., Sonsupap, S., Maensiri, S. & Kidkhunthod, P. Room-temperature ferromagnetism in Fe-doped In2O3 nanoparticles. App. Phys. A 121, 239–244 (2015).
Daengsakul, S. et al. The effect of Gd doping in La1−x−yGdxSryMnO3 compound on nanocrystalline structure by X-ray Absorption Spectroscopy (XAS) technique. Microelectron. Eng. 146, 38–42 (2015).
Ghaemi, M., Ataherian, F., Zolfaghari, A. & Jafari, S. M. Charge storage mechanism of sonochemically prepared MnO2 as supercapacitor electrode: Effects of physisorbed water and proton conduction. Electrochimica Acta 53, 4607–4614 (2008).
Simon, P. & Gogotsi, Y. Materials for electrochemical capacitors. Nat. Mater. 7, 845–854 (2008).
Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung dünner Schichten und zur Mikrowägung. Z. Phys. 155, 206–222 (1959).
Tsai, W.-Y., Taberna, P.-L. & Simon, P. Electrochemical Quartz Crystal Microbalance (EQCM) Study of Ion Dynamics in Nanoporous Carbons. J. Am. Chem. Soc. 136, 8722–8728 (2014).
Yan, J. et al. Advanced Asymmetric Supercapacitors Based on Ni(OH)2/Graphene and Porous Graphene Electrodes with High Energy Density. Adv. Funct. Mater. 22, 2632–2641 (2012).
Hummers, W. S. & Offeman, R. E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 80, 1339–1339 (1958).
Luanwuthi, S., Krittayavathananon, A., Srimuk, P. & Sawangphruk, M. In situ synthesis of permselective zeolitic imidazolate framework-8/graphene oxide composites: rotating disk electrode and Langmuir adsorption isotherm. RSC Adv. 5, 46617–46623 (2015).
Sanguansak, Y. et al. Permselective properties of graphene oxide and reduced graphene oxide electrodes. Carbon 68, 662–669 (2014).
Krittayavathananon, A., Iamprasertkun, P. & Sawangphruk, M. Enhancing the charge-storage performance of N-doped reduced graphene oxide aerogel supercapacitors by adsorption of the cationic electrolytes with single-stand deoxyribonucleic acid. Carbon 109, 314–320 (2016).
McDonough, J. K. et al. Influence of the structure of carbon onions on their electrochemical performance in supercapacitor electrodes. Carbon 50, 3298–3309 (2012).
Lu, Y. et al. Hierarchical, porous CuS microspheres integrated with carbon nanotubes for high-performance supercapacitors. Scientific Reports 5, 16584 (2015).
Yang, J. et al. Rapid and controllable synthesis of nitrogen doped reduced graphene oxide using microwave-assisted hydrothermal reaction for high power-density supercapacitors. Carbon 73, 106–113 (2014).
Singh, A. & Chandra, A. Significant Performance Enhancement in Asymmetric Supercapacitors based on Metal Oxides, Carbon nanotubes and Neutral Aqueous Electrolyte. Sci. Rep. 5, 15551 (2015).
Tooming, T., Thomberg, T., Kurig, H., Jänes, A. & Lust, E. High power density supercapacitors based on the carbon dioxide activated d-glucose derived carbon electrodes and 1-ethyl-3-methylimidazolium tetrafluoroborate ionic liquid. J. Power Sources 280, 667–677 (2015).
Berton, N. et al. Wide-voltage-window silicon nanowire electrodes for micro-supercapacitors via electrochemical surface oxidation in ionic liquid electrolyte. Electrochem. Commun. 41, 31–34 (2014).
Chu, A. & Braatz, P. Comparison of commercial supercapacitors and high-power lithium-ion batteries for power-assist applications in hybrid electric vehicles: I. Initial characterization. J. Power Sources 112, 236–246 (2002).
Xiao, X. et al. Freestanding functionalized carbon nanotube-based electrode for solid-state asymmetric supercapacitors. Nano Energy 6, 1–9 (2014).
Huang, P.-L. et al. Ionic Liquid Electrolytes with Various Constituent Ions for Graphene-based Supercapacitors. Electrochim. Acta 161, 371–377 (2015).
Acknowledgements
Financial supports by Thailand Research Fund (TRF) and Vidyasirimedhi Institute of Science and Technology (VISTEC) are acknowledged (RSA5880043). This research is also supported in part by the Graduate Program Scholarship from the Graduate School, Kasetsart University. Supports from the Center of Excellence on Petrochemical and Materials Technology (PETROMAT), the Kasetsart University Research and Development Institute (KURDI), Department of Chemical Engineering, Kasetsart University, National Research University Project of Thailand (NRU), Synchrotron Light Research Institute (BL5.1 and BL5.2) (Public Organization), Thailand for XANES and XPS facilities, and the Frontier Research Center at VISTEC are also acknowledged.
Author information
Authors and Affiliations
Contributions
P.I. and A.K. synthesized the materials and performed the electrochemical experiment and discussed the results. M.S. designed and directed the work, discussed the results, and wrote the manuscript. N.C., P.K., W.S., S.N., P.P., and S.I. helped for the sample measurements including FTIR, RAMAN, XPS, XAS, SEM and TEM. A.S., S.M., R.Y., K.K., and J.L. discussed the results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Iamprasertkun, P., Krittayavathananon, A., Seubsai, A. et al. Charge storage mechanisms of manganese oxide nanosheets and N-doped reduced graphene oxide aerogel for high-performance asymmetric supercapacitors. Sci Rep 6, 37560 (2016). https://doi.org/10.1038/srep37560
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37560
This article is cited by
-
Enhanced electrochemical energy storage devices utilizing a one-dimensional (1D) α-MnO2 nanocomposite encased in onion-like carbon
Journal of Materials Science (2024)
-
Scalable fabrication of hierarchically porous N-doped carbon electrode materials for high-performance aqueous symmetric supercapacitor
Journal of Materials Science (2018)
-
Nitrogen-doped porous 3D graphene with enhanced supercapacitor properties
Journal of Materials Science (2018)
-
Graphene/Polyaniline Aerogel with Superelasticity and High Capacitance as Highly Compression-Tolerant Supercapacitor Electrode
Nanoscale Research Letters (2017)
-
Three-Dimensional Bi-Continuous Nanoporous Gold/Nickel Foam Supported MnO2 for High Performance Supercapacitors
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
