
Abstract
The unicellular alga Dunaliella bardawil is a highly salt-tolerant organism, capable of accumulating glycerol, glycine betaine and β-carotene under salt stress, and has been considered as an excellent model organism to investigate the molecular mechanisms of salt stress responses. In this study, several carotenogenic genes (DbCRTISO, DbZISO, DbLycE and DbChyB), DbBADH genes involved in glycine betaine synthesis and genes encoding probable WRKY transcription factors from D. bardawil were isolated, and promoters of DbCRTISO and DbChyB were cloned. The promoters of DbPSY, DbLycB, DbGGPS, DbCRTISO and DbChyB contained the salt-regulated element (SRE), GT1GMSCAM4, while the DbGGPS promoter has another SRE, DRECRTCOREAT. All promoters of the carotenogenic genes had light-regulated elements and W-box cis-acting elements. Most WRKY transcription factors can bind to the W-box, and play roles in abiotic stress. qRT-PCR analysis showed that salt stress up-regulated both carotenogenic genes and WRKY transcription factors. In contrast, the transcription levels of DbBADH showed minor changes. In D. bardawil, it appears that carotenoid over-accumulation allows for the long-term adaptation to salt stress, while the rapid modulation of glycine betaine biosynthesis provides an initial response.
Similar content being viewed by others

Introduction
Research on Dunaliella bardawil has mainly focused on its salinity tolerance and high β-carotene accumulation under various abiotic stresses (high light, high salinity or nutrient limitation)1,2. Exploring the stress-induced carotenoid metabolism and elucidating the mechanisms of stress responses and regulation in D. bardawil are important for the development of β-carotene production. The carotenoid biosynthesis pathway of D. bardawil has been well described by Liang et al.3. Geranylgeranyl diphosphate (GGPP), the C20 precursor of phytoene, can be synthesized by geranylgeranyl pyrophosphate synthase (GGPS) from three IPP molecules and one DMAPP molecule. The condensation of two GGPP molecules produces phytoene (C40), catalyzed by phytoene synthase (PSY). Then phytoene is desaturated by phytoene desaturases (PDS) and ζ-carotene desaturases (ZDS) and isomerized by 15-cis-ζ-carotene isomerase (ZISO) and carotenoid isomerase (CRTISO) to form the linear all-trans lycopene. The cyclation of lycopene by lycopene ε-cyclase (LycE) and lycopene β-cyclase (LycB) introduces ε- and β-ionone end groups, respectively, yielding α-carotene and β-carotene. β-carotene is hydroxylated by β–carotene hydroxylase (ChyB) to produce zeaxanthin.
D. bardawil is an impressively salt-tolerant organism, and can adapt to hypersaline environments of up to 5.5 M NaCl. The adaptation of D. bardawil to salt stress is attributed to an initial rapid accumulation of glycerol and glycine betaine as intracellular osmoprotectants4. Subsequently, the long-term response mechanisms, including salt-induced genes expression, salt tolerance related proteins or transcription factors, allow D. bardawil cells to acclimate to high salinity2. The regulation of genes related to glycerol metabolism in Dunaliella salina under high salt stress conditions has been investigated5, and it was suggested that dihydroxyacetone reductase may be a key enzyme in the regulation of glycerol metabolism under salt stress. However, genes involved in glycine betaine metabolism have not yet been studied. In the chloroplast of higher plants, glycine betaine is synthesized from serine via ethanolamine, choline and betaine aldehyde; betaine aldehyde dehydrogenase (BADH) is an important enzyme in the process6. Glycine betaine can be accumulated during stress response in many crop plants, including sugarbeet, spinach, barley, wheat and sorghum. However, several plants such as rice, rockcress, tobacco, and potato, naturally cannot synthesize glycine betaine under either stress or non-stress conditions7. It has been reported that overexpression of spinach BADH in sweet potato (Ipomoea batatas) can improve tolerance to various abiotic stresses, including salt, oxidative stress, and low temperature8. Thus it is of interest to study BADH from D. bardawil to reveal the mechanism of salt tolerance and provide tools to potentially engineer salt-tolerant.crop plants.
To investigate the molecular mechanism of salt tolerance in Dunaliella, proteomics technology has been used to identify the salt-induced proteins. Dunaliella responds to salt stress by improving photosynthetic CO2 assimilation and directing carbon and energy resources to glycerol biosynthesis9. Another report found that the major salt-regulated proteins were involved in membrane structure stabilization and signal transduction pathways in D. salina10. Changes in multiple biological process (such as protein synthesis, membrane stabilization, signal transduction, and redox energy production) in response to salt stress suggested that more than one mechanism may play a role in the unique capacity of salt tolerance of Dunaliella cells2.
It has been reported that cis-acting elements and transcription factors also play important roles in plant abiotic stresses11,12. cis-Acting elements exist in the vicinity of the structural portion of a gene, and typically regulate gene transcription by functioning as binding sites for transcription factors. In plants, transcription factors, such as basic leucine-zipper (bZIP), MYB-type, dehydration-responsive-element binding (DREB) proteins and WRKY transcription factors, have been identified and the roles of these transcription factors are still being studied11. In algae, MYB-type transcription factors were found to regulate CO2-responsive genes and phosphate starvation signaling in Chlamydomonas reinhardtii13,14. In D. bardawil, the stress response genes DbPSY, DbPDS, DbZDS and DbLycB have been isolated, and the cis-acting elements in these gene promoters analyzed15,16.
Recently, the transcriptome sequencing and annotation of Dunaliella tertiolecta UTEX LB 999 has been reported17, which provides a valuable resource for research on genes encoding key enzymes involved in carotenoid biosynthesis and salt stress regulation. In this study, we investigated the mechanisms of carotenoid accumulation during salt stress in D. bardawil. With the help of transcriptome sequencing, key genes (DbCRTISO, DbZISO, DbLycE and DbChyB) involved in carotenoid biosynthesis, the BADH gene involved in glycine betaine biosynthesis, and genes encoding probable WRKY transcription factors were isolated. The transcription levels of these genes were analyzed by quantitative Real-Time PCR (qRT-PCR) during salt stress. The promoters of DbCRTISO and DbChyB were cloned by genome walking, and the cis-acting elements related to salt regulation were analyzed.
Materials and Methods
Algal strains and cultivation conditions
The green alga D. bardawil strain FACHB-847 was obtained from the Institute of Hydrobiology, Chinese Academy of Science. D. bardawil cells were grown in a defined medium16 at 26 °C under a 16/8 h light/dark cycle for 7~10 days. In order to study salt stress in D. bardawil, algal cells were grown in the defined medium with 0.5 M, 1.0 M, 1.5 M, 2.0 M, 2.5 M, 3.0 M, 3.5 M and 4.0 M NaCl.
Transcriptome sequencing and CDSs identification
The transcriptome sequencing and annotation of Dunaliella tertiolecta UTEX LB 999 (NCBI accession number: SRA023642) has been reported17; this can provide a valuable resource for research on genes encoding key enzymes involved in carotenoids biosynthesis and salt stress regulation in this study. We downloaded the two documents SRR070441 and SRR070442 of SRA023642 in NCBI SRA database. The RNA sequencing reads of D. tertiolecta UTEX LB 999 were assembled and clustered into 35,625 unigenes. These unigenes were aligned to three public protein databases (Nr, Swiss-Prot and KEGG) by blastx. The Blast2GO software was used to obtain GO annotations for the unigenes. Several coding sequences (CDSs) encoding key enzymes involved in carotenoid biosynthesis (related enzymes like CRTISO, ZISO, LycE and ChyB) and salt stress regulation (related proteins like BADH and WRKY transcription factors) were identified in these unigenes. We used this sequence data to screen D. bardawil for related sequences through PCR amplification (testing primers shown in Supplemental Table S1), subcloning and sequencing.
Genomic DNA isolation and promoters cloning
Genomic DNA extraction from D. bardawil in the log or late log phase was performed according to the OMEGA HP Plant DNA Kit. Genome walking was implemented with genome walking kit (Takara). Based on the user manual of the genome walking kit, gene-specific primers (Table 1) were designed to isolate the promoters of DbCRTISO and DbChyB.
Bioinformatics Analysis
Sequence similarity analysis was performed using BLAST (http://blast.ncbi.nlm.nih.gov/). The sequence alignment was generated using ClustalX2 software. Conserved domains within a protein or coding nucleotide sequence were detected using the Conserved Domains Search tool (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Translation prediction was performed using the Translation tool (http://web.expasy.org/translate/). The open reading frames (ORFs) in a nucleotide sequence were predicted by ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). Gene structure was displayed by GSDS2.0 (http://gsds.cbi.pku.edu.cn/). Promoter analysis was performed with the PLACE (https://sogo.dna.affrc.go.jp/cgi-bin/sogo.cgi?lang=en&pj=640&action=page&page=newplace) and PlantCARE websites (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). The database of plant transcription factors is available at the PlantTFDB website (http://planttfdb.cbi.pku.edu.cn/).
Total RNA extraction and Quantitative Real-Time PCR (qRT-PCR) analysis
The total RNAs were prepared from D. bardawil cells cultivated with 0.5 M, 1.0 M, 1.5 M, 2.0 M, 2.5 M, 3.0 M, 3.5 M and 4.0 M NaCl in the late log phase using RNAiso Plus Reagent (Takara). RT-PCR was performed with a 7500 Real-Time PCR System (Applied Biosystems) using the PrimeScript RT Reagent Kit with gDNA Eraser and the SYBR Green PCR Kit (Takara). Primers for quantitative analysis were designed according to the identified CDSs of the corresponding genes and listed in Supplemental Table S2. All PCR reactions were set up in triplicate, and every sample was replicated in parallel three times to ensure statistical relevance. The PCR conditions were used as followed: 30 s at 95 °C, and then 40 cycles of 5 s at 95 °C and 34 s at 60 °C. The specificity of qRT-PCR primers was monitored by the presence of dissociation curves with single peaks and sequencing of the corresponding products with unique bands of the expected sizes. Data were collected and analyzed using SDS software (Applied Biosystems). All results were normalized to the glyceraldehyde-3-phosphate dehydrogenase gene from D. bardawil (DbGAPDH).
Results
Transcriptome analysis and CDSs identification from D. bardawil
After PCR amplification, subcloning and sequencing, we found that the coding sequences (CDS) of the genes of interest from D. bardawil FACHB-847 were 100% identical to those from D. tertiolecta UTEX LB 999. The CDS of all these genes were identified and the details were shown in Table 2. The 1488 bp full-length CDS of DbCRTISO encoded 495 Aa, which displayed 60% and 59% sequence similarity with those of C. reinhardtii and Haematococcus pluvialis, respectively. The partial CDS of DbZISO was identified, and the deduced amino acid sequences showed 64% and 62% identities with those of Ceratophyllum demersum and Arabidopsis thaliana, respectively. Using ORF Finder tool, the Unigene0014471 contained a 1314 bp putative ORF and the Unigene0012559 contained a 954 putative ORF. The 1314 bp putative ORF encoded part of lycopene epsilon cyclase (LycE), with sequence identities of 69% and 64% with those of H. pluvialis and C. reinhardtii, respectively. The 954 bp putative ORF encoding partial β-carotene hydroxylase (ChyB), displayed 70% and 56% similarities with those of D. salina and C. reinhardtii, respectively. Previously in our research group, genes such as GGPS, PSY, PDS, ZDS and LycB have been isolated from D. bardawil FACHB-8473,15,18.
A 1496 bp partial DbBADH was identified, as the deduced amino acid sequences showed 56% and 55% identities with those of Triticum aestivum and Glycine max, respectively. According to transcriptome annotations, five unigenes encoding probable WRKY transcription factors were found, but four unigenes were identified (Table 2). Because one unigene putatively encoding the expected DbWRKY5 cannot be cloned according to the testing primers in Supplemental Table S1.
The features of the probable WRKY transcription factors from D. bardawil
WRKY transcription factors play roles in many processes in plants, including regulating many stress reactions19. They contain either one or two WRKY protein domains. The WRKY protein domain is a DNA binding domain, about 60 amino acids in length, which contains a highly conserved core WRKYGQ(E)K motif and a zinc-finger region. The cysteine and histidine zinc-finger domain is either CX4-5CX22-23HXH or CX7CX23HXC, where X means any amino acid20. Most WRKY transcription factors can specifically bind to the W-box cis-regulatory element that has a consensus sequence of TTGACC/T20.
Here, four probable WRKY transcription factors were identified from D. bardawil, and defined as DbWRKY1, DbWRKY2, DbWRKY3 and DbWRKY4. The conserved domains of the four putative amino acid sequences were predicted by Conserved Domains Search and shown in Fig. 1A. All of them had the WRKYGEK signature motif and a zinc-finger structure at the C-terminus (Fig. 1B). The zinc-finger structure was CX4CX23HXH in DbWRKY1, CX4CX21HXH in DbWRKY2, and CX4CX22HXH in DbWRKY3 and DbWRKY4.

(A) Conserved domain analysis of four probable WRKY transcription factors from D. bardawil. (B) Alignment of the four WRKY protein domains from D. bardawil by ClustalX2. The WRKY motif is highlighted in a red frame and the cysteines and histidines that form the zinc-finger structures are shown in red triangles.
Promoter cloning and genomic structures of carotenogenic genes
We used the full-length CDSs of DbCRTISO and DbChyB to identify the promoter regions and genomic DNA of DbCRTISO and DbChyB by genome walking. Sequence analysis by BlastN found that the nucleotide sequence of the third nested PCR product (Fig. 2) possesses identical regions (51 bp in length) as expected with the 5′-DbCRTISO, and 37 bp in length with the 5′-DbChyB., defining the promoter regions of DbCRTISO and DbChyB to be 2118 bp and 2487 bp in length, respectively.

(A) Promoter isolation of DbCRTISO by genome walking. (B) Promoter isolation of DbChyB by genome walking. Lane M, 500 bp DNA ladder marker; lanes 1-3, products of 1st PCR, 2nd nested PCR, and 3rd nested PCR, respectively.
Analysis of the nucleotide sequences of the cloned genomic DNA fragment indicated that DbCRTISO contains 16 exons interrupted by 15 introns, and DbChyB has 7 exons and 6 introns (Fig. 3). It appears that the genomic structures of genes from D. bardawil are much more complicated (more intron numbers and longer average intron length) than that of the corresponding genes from other algae and plants (Fig. 3). The full-length cDNA and the genomic DNA of DbGGPS have been isolated; DbGGPS contained 10 exons interrupted by 9 introns and the cloned DbGGPS promoter region was 871 bp in length3. In our research group, the promoter regions of PSY, PDS, ZDS and LycB from D. bardawil have also been cloned15,16.

(A) Distribution of exons and introns in the genomic DNA of CRTISOs. The sequences used for analysis are as follows: CRTISO from green algae: DbCRTISO (in this study); CrCRTISO, Chlamydomonas reinhardtii (NW_001843787.1); CsCRTISO, Coccomyxa subellipsoidea C-169 (NW_005178040.1); CRTISO from diatoms: TpCRTISO, Thalassiosira pseudonana (NC_012070.1); PtCRTISO, Phaeodactylum tricornutum (NC_011675.1); CRTISO from red algae: GsCRTISO, Galdieria sulphuraria (NW_005178468.1); CmCRTISO, Cyanidioschyzon merolae (NC_010140.1); CRTISO from higher plants: AtCRTISO, Arabidopsis thaliana (NC_003070.9); ZmCRTISO, Zea mays (NC_024462.1). (B) Distribution of exons and introns in the genomic DNA of ChyBs. The sequences used for analysis are as follows: ChyB from green algae: DbChyB (in this study); CrChyB, Chlamydomonas reinhardtii (NW_001843792.1); BpChyB, Bathycoccus prasinos (NC_023998.1); MpChyB, Micromonas pusilla CCMP1545 (NW_003315882.1); VcChyB, Volvox carteri f. nagariensis (NW_003307618.1); ChyB from higher plants: AtChyB, Arabidopsis thaliana (NW_003302549.1); CsChyB, Citrus sinensis cultivar Valencia (NC_023054.1); SlChyB, Solanum lycopersicum cultivar Heinz 1706 (NC_015440.2); ZmChyB, Zea mays (NC_024468.1).
cis-Acting element analysis
Presently, the promoters of seven genes (DbPSY, DbPDS, DbZDS, DbLycB, DbGGPS, DbCRTISO and DbChyB) involved in carotenoid metabolism in D. bardawil have been isolated3,15,16. cis-Acting elements of DbPSY, DbPDS, DbZDS and DbLycB promoters have been analyzed15,16. Here we comprehensively analyzed several stress-responsive cis-acting elements of the seven gene promoters by PLACE and PlantCARE and the results are shown in Table 3. Promoters of DbPSY, DbLycB, DbGGPS, DbCRTISO and DbChyB genes contained GT1GMSCAM4, salt-regulated elements (SREs), defined as cis-acting elements that can control NaCl-induced expression or salt-responsive gene expression. The DbPDS promoter did not have any SREs. As well, the DbGGPS promoter contained the DRECRTCOREAT motif involved in drought-, high-salt- and cold-responsive gene expression. Exceptionally, only the DbZDS promoter contained the hypoosmolarity-responsive element (HRE)16. Promoters of the DbPSY, DbZDS, DbGGPS, DbCRTISO and DbChyB genes contained ASF1MOTIFCAMV, involved in abiotic and biotic stress. All seven of the gene promoters contained light-regulated elements and W-boxes (which might be recognized by WRKY transcription factors).
qRT-PCR analysis of genes in response to salt stress in D. bardawil
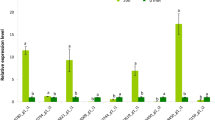
The transcription levels of genes involved in carotenoid biosynthesis, glycine betaine biosynthesis and encoding WRKY transcription factors, were analyzed by qRT-PCR during salt stress. For the carotenoid biosynthesis pathway under salt stress (Fig. 4A), six carotenogenic genes (DbGGPS, DbPSY, DbZISO, DbLycB, DbLycE and DbChyB) were up-regulated, while the relative expression levels of three carotenogenic genes (DbPDS, DbZDS and DbCRTISO) were increased slightly. For the glycine betaine biosynthesis pathway under salt stress (Fig. 4B), the relative expression levels of DbBADH were increased steadily with the NaCl concentration (from 0.5 M to 2.0 M), and then decreased slightly in the treatments from 2.0 M to 4.0 M NaCl. In addition, it was shown that genes encoding the four probable WRKY transcription factors were up-regulated under salt stress (Fig. 4B).

(A) qRT-PCR analysis of the transcription levels of genes involved in carotenoid biosynthesis. (B) qRT-PCR analysis of the transcription levels of genes involved in glycine betaine biosynthesis and encoding WRKY transcription factors.
Discussion
The transcriptome sequencing and annotation of D. tertiolecta UTEX LB 999, provides a valuable resource for research on genes encoding key enzymes involved in carotenoids biosynthesis and salt stress regulation. Recently, the ZISO gene was identified in maize21. It was confirmed that in higher plants two isomerases (CRTISO and ZISO) are involved in the isomerization that synthesizes all-trans lycopene from phytoene21,22. In this study, these two isomerases were also found in D. bardawil, which gave us a new insight into carotenoid metabolism in D. bardawil, a halotolerant alga. Currently, a genome sequencing project for Dunaliella salina strain CCAP 19/18 is in process2. The availability of more genomic data of Dunaliella will provide us with a better understanding of the genomic structures of the carotenogenic genes, their gene regulation and their molecular evolution. Presently, we have isolated the genomic DNA of DbPSY (6 exons interrupted by 5 introns)15, DbZDS (12 exons interrupted by 11 introns)16, DbGGPS (10 exons interrupted by 9 introns)3, DbCRTISO (16 exons interrupted by 15 introns, Fig. 3A), DbChyB (7 exons interrupted by 6 introns, Fig. 3B), and found the genomic structures of these genes were more complex than that from other species. In addition, two subunits of ATP-citrate lyase from Dunaliella tertiolecta (DtACLA and DtACLB) were isolated, and there were 12 exons and 11 introns in DtACLA, and 20 exons and 19 introns in DtACLB23. The gene DsHsp90 from D. salina encoding heat shock protein 90 contained 21 exons interrupted by 20 introns24. It appears that genes from Dunaliella may be intron-rich24.
In various biological processes, including abiotic stress responses, hormone responses and developmental processes, the regulation of gene transcription initiation and accurate transcription efficiency can be controlled by cis-acting elements in the promoter regions of stress-inducible genes and transcription factors25. It appears that several carotenogenic genes (DbPSY, DbLycB, DbGGPS, DbCRTISO, DbChyB) in D. bardawil were salt stress-inducible as the promoter regions of these genes contained one or more salt-regulated elements (SREs), such as GT1GMSCAM4 and DRECRTCOREAT (Table 3). In addition, salt stress up-regulated most carotenogenic genes in D. bardawil, except for DbZDS and DbCRTISO (Fig. 4A).
In Haematococcus pluvialis strains from Queensland, the transcript abundance of seven carotenogenic genes [IPI-1, IPI-2, PSY, LycB, crtR-B (encoding β-ring hydroxylase), BKT2 (encoding β-carotene ketolase), and crtO (β-carotene oxygenase)] was up-regulated upon salinity stress26. And under nutrient stress conditions, carotenogenic genes of H. pluvialis, such as PSY, PDS, LycB, BKT, and ChyB were also up-regulated27. In C. reinhardtii, PSY and PDS displayed a rapid up-regulation in response to light28, suggesting that up-regulation of carotenoid biosynthetic pathway genes may occur in response to abiotic stress environments in algae.
All the carotenogenic genes we studied contained the W-box cis-acting elements, which can be recognized by WRKY transcription factors. Recently, it has been reported that WRKY transcription factors play key roles in regulating many stress responses in plants19. The Plant Transcription Factor Database (PlantTFDB) shows that many more WRKY transcription factors are identified in higher plants than in green algae. There are over 80 WRKY genes in Arabidopsis thaliana29 and Oryza sativa30. In green algae, such as Chlamydomonas reinhardtii, Ostreococcus sp. RCC809 and Volvox carteri, the number of WRKY transcription factors varies from 1 to 3. The evolution of the WRKY transcription factor family between green algae and land plants has been reported31. In this study, four probable WRKY transcription factors were identified, and they were up-regulated under salt stress (Fig. 4B). The binding of WRKY transcription factors to W-boxes is a feature of both biotic and abiotic stress responses. It seemed that these four WRKY transcription factors might act as activators under salt stress. These four WRKY transcription factors can be further studied by overexpression in higher plants; it was reported that overexpression of OsWRKY45 can lead to increased salt and drought tolerance, and enhanced disease resistance32, and in Arabidopsis, overexpression of either AtWRKY25 or AtWRKY33 can result in increased salt tolerance33. Intriguingly, compared with the carotenogenic genes, the transcription level changes of DbBADH under salt stress were small (Fig. 4B). It appears that carotenoid over-accumulation allows for long-term adaptation to salt stress in D. bardawil, and the rapid adjustment of glycine betaine biosynthesis occurs at the start of the salt stress2,4. The adaptation of D. bardawil to salt stress can thus be divided into two stages: first, rapid adjustment of the intracellular concentration of glycerol and glycine betaine, as intracellular osmoprotectants; and second, a long-term response that might include carotenoid accumulation2.
Taken together, in D. bardawil, promoter regions of stress-inducible genes involved in carotenoid biosynthesis contain cis-acting elements, such as SRE, DRE, light-regulated elements, involved in stress-responsive gene expression. Various transcription factors are involved in the regulation of stress-inducible genes. The analysis of cis-acting elements and WRKY transcription factors in D. bardawil can give us a better understanding of regulatory mechanisms involved in salt stress-responsive gene expression.
Additional Information
How to cite this article: Liang, M.-H. and Jiang, J.-G. Analysis of carotenogenic genes promoters and WRKY transcription factors in response to salt stress in Dunaliella bardawil. Sci. Rep. 7, 37025; doi: 10.1038/srep37025 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Ben-Amotz, A., Katz, A. & Avron, M. Accumulation of β-carotene in halotolerant alge: purification and characterization of β-carotene-rich globules from Dunaliella bardawil (chlorophyceae). J. Phycol. 18, 529–537 (1982).
Ramos, A. A. et al. The unicellular green alga Dunaliella salina Teod. as a model for abiotic stress tolerance: genetic advances and future perspectives. Algae 26, 3–20 (2011).
Liang, M.-H., Liang, Y.-J., Jin, H.-H. & Jiang, J.-G. Characterization and Functional Identification of a Gene Encoding Geranylgeranyl Diphosphate Synthase (GGPS) from Dunaliella bardawil . J. Agric. Food Chem. 63, 7805–7812 (2015).
Mishra, A., Mandoli, A. & Jha, B. Physiological characterization and stress-induced metabolic responses of Dunaliella salina isolated from salt pan. J. Ind. Microbiol. Biotechnol. 35, 1093–1101 (2008).
Zhao, L. n., Gong, W. f., Chen, X. w. & Chen, D. f. Characterization of genes and enzymes in Dunaliella salina involved in glycerol metabolism in response to salt changes. Phycol. Res. 61, 37–45 (2013).
Ashraf, M. & Foolad, M. Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ. Exp. Bot. 59, 206–216 (2007).
Sakamoto, A. & Murata, N. Genetic engineering of glycinebetaine synthesis in plants: current status and implications for enhancement of stress tolerance. J. Exp. Bot. 51, 81–88 (2000).
Fan, W., Zhang, M., Zhang, H. & Zhang, P. Improved tolerance to various abiotic stresses in transgenic sweet potato (Ipomoea batatas) expressing spinach betaine aldehyde dehydrogenase. PLoS One 7, e37344 (2012).
Liska, A. J., Shevchenko, A., Pick, U. & Katz, A. Enhanced photosynthesis and redox energy production contribute to salinity tolerance in Dunaliella as revealed by homology-based proteomics. Plant Physiol. 136, 2806–2817 (2004).
Katz, A., Waridel, P., Shevchenko, A. & Pick, U. Salt-induced changes in the plasma membrane proteome of the halotolerant alga Dunaliella salina as revealed by blue native gel electrophoresis and nano-LC-MS/MS analysis. Mol. Cell. Proteomics 6, 1459–1472 (2007).
Saibo, N. J., Lourenço, T. & Oliveira, M. M. Transcription factors and regulation of photosynthetic and related metabolism under environmental stresses. Ann. Bot. 103, 609–623 (2009).
Chinnusamy, V., Zhu, J. & Zhu, J.-K. Salt stress signaling and mechanisms of plant salt tolerance. Genet. Eng. 27, 141–177 (2006).
Yoshioka, S. et al. The novel Myb transcription factor LCR1 regulates the CO2-responsive gene Cah1, encoding a periplasmic carbonic anhydrase in Chlamydomonas reinhardtii . Plant Cell 16, 1466–1477 (2004).
Rubio, V. et al. A conserved MYB transcription factor involved in phosphate starvation signaling both in vascular plants and in unicellular algae. Genes Dev. 15, 2122–2133 (2001).
Lao, Y.-M., Xiao, L., Jiang, J.-G. & Zhou, S.-S. In silico analysis of phytoene synthase and its promoter reveals hints for regulation mechanisms of carotenogenesis in Duanliella bardawil . Bioinformatics 27, 2201–2208 (2011).
Lao, Y.-M., Xiao, L., Luo, L.-X. & Jiang, J.-G. Hypoosmotic Expression of Dunaliella bardawil ζ-Carotene Desaturase Is Attributed to a Hypoosmolarity-Responsive Element Different from Other Key Carotenogenic Genes. Plant Physiol. 165, 359–372 (2014).
Rismani-Yazdi, H., Haznedaroglu, B. Z., Bibby, K. & Peccia, J. Transcriptome sequencing and annotation of the microalgae Dunaliella tertiolecta: pathway description and gene discovery for production of next-generation biofuels. BMC Genomics 12, 148 (2011).
Ye, Z.-W., Liu, G.-N. & Jiang, J.-G. Structural and phylogenetic analysis of a novel ζ‐carotene desaturase from Dunaliella bardawil, a unicellular alga that accumulates large amounts of β‐carotene. Limnol. Oceanogr. 56, 133–138 (2011).
Chen, L. et al. The role of WRKY transcription factors in plant abiotic stresses. Biochim. Biophys. Acta, Gene Regul. Mech. 1819, 120–128 (2012).
Rushton, P. J., Somssich, I. E., Ringler, P. & Shen, Q. J. WRKY transcription factors. Trends Plant Sci. 15, 247–258 (2010).
Li, F., Murillo, C. & Wurtzel, E. T. Maize Y9 encodes a product essential for 15-cis-ζ-carotene isomerization. Plant Physiol. 144, 1181–1189 (2007).
Nisar, N., Li, L., Lu, S., Khin, N. C. & Pogson, B. J. Carotenoid metabolism in plants. Mol. Plant 8, 68–82 (2015).
Liang, M.-H. & Jiang J.-G. Characterization and nitrogen deficiency response of ATP-citrate lyase from unicellular alga Dunaliella tertiolecta. Algal Research 20, 77–86 (2016).
Wang, S.-J., Wu, M.-J., Chen, X.-J., Jiang, Y. & Yan, Y.-B. DsHsp90 Is Involved in the Early Response of Dunaliella salina to Environmental Stress. Int. J. Mol. Sci. 13, 7963–7979 (2012).
Yamaguchi-Shinozaki, K. & Shinozaki, K. Organization of cis-acting regulatory elements in osmotic-and cold-stress-responsive promoters. Trends Plant Sci. 10, 88–94 (2005).
Gao, Z. et al. Comparison of astaxanthin accumulation and biosynthesis gene expression of three Haematococcus pluvialis strains upon salinity stress. J. Appl. Phycol. 27, 1853–1860 (2015).
Vidhyavathi, R., Venkatachalam, L., Sarada, R. & Ravishankar, G. A. Regulation of carotenoid biosynthetic genes expression and carotenoid accumulation in the green alga Haematococcus pluvialis under nutrient stress conditions. J. Exp. Bot. 59, 1409–1418 (2008).
Bohne, F. & Linden, H. Regulation of carotenoid biosynthesis genes in response to light in Chlamydomonas reinhardtii . Biochim. Biophys. Acta, Gene Struct. Expression 1579, 26–34 (2002).
Eulgem, T., Rushton, P. J., Robatzek, S. & Somssich, I. E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5, 199–206 (2000).
Wu, K.-L., Guo, Z.-J., Wang, H.-H. & Li, J. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res. 12, 9–26 (2005).
Rinerson, C. I., Rabara, R. C., Tripathi, P., Shen, Q. J. & Rushton, P. J. The evolution of WRKY transcription factors. BMC Plant Biol. 15, 66 (2015).
Qiu, Y. & Yu, D. Over-expression of the stress-induced OsWRKY45 enhances disease resistance and drought tolerance in Arabidopsis . Environ. Exp. Bot. 65, 35–47 (2009).
Jiang, Y. & Deyholos, M. K. Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 69, 91–105 (2009).
Acknowledgements
This study was funded by the National Natural Foundation of China (31171631 and 31571773), Guangdong Province Science and technology plan project (2011B031200005), and Guangdong Provincial Bureau of ocean and fishery science and technology to promote a special (A201301C04).
Author information
Authors and Affiliations
Contributions
M.L. performed the experiments and wrote the manuscript, and J.J. designed the experiments and revised the manuscript. All authors discussed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liang, MH., Jiang, JG. Analysis of carotenogenic genes promoters and WRKY transcription factors in response to salt stress in Dunaliella bardawil. Sci Rep 7, 37025 (2017). https://doi.org/10.1038/srep37025
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37025
This article is cited by
-
Comparative morphological, physiological, biochemical and genomic studies reveal novel genes of Dunaliella bioculata and D. quartolecta in response to salt stress
Molecular Biology Reports (2022)
-
De novo transcriptome sequencing and analysis of salt-, alkali-, and drought-responsive genes in Sophora alopecuroides
BMC Genomics (2020)
-
Transcriptome-wide identification of WRKY transcription factor and their expression profiles under salt stress in sweetpotato (Ipomoea batatas L.)
Plant Biotechnology Reports (2020)
-
Identification of key genes and regulators associated with carotenoid metabolism in apricot (Prunus armeniaca) fruit using weighted gene coexpression network analysis
BMC Genomics (2019)
-
Comparative transcriptomics approach in elucidation of carotenoid biosynthesis regulation in grains of rice (Oryza sativa L.)
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
