
Abstract
Phospholipase D1 (PLD1) plays a known role in several differentiation processes, but its role in adipogenic differentiation remains unknown. In the present study, we identified PLD1 as a negative regulator of adipogenic differentiation. We showed that PLD activity was downregulated by both 3-Isobutyl-1-methylxanthine (IBMX) and insulin upon induction of differentiation in 3T3-L1 adipogenic cells. In line with this observation, PLD activity decreased in both high fat diet (HFD)-fed mice and ob/ob mice. We also found that differentiation of 3T3-L1 preadipocytes was enhanced by the depletion of PLD1 levels or inhibition of PLD1 activity by VU0155069, a PLD1-specific inhibitor. Conversely, treatment with phosphatidic acid (PA), a PLD product, and overexpression of PLD1 both caused a decrease in adipogenic differentiation. Moreover, the elevated differentiation in PLD1-knockdown 3T3-L1 cells was reduced by either PA treatment or PLD1 expression, confirming negative roles of PLD1 and PA in adipogenic differentiation. Further investigation revealed that PA displaces DEP domain-containing mTOR-interacting protein (DEPTOR) from mTORC1, which subsequently phosphorylates insulin receptor substrate-1 (IRS-1) at serine 636/639 in 3T3-L1 cells. Taken together, our findings provide convincing evidence for a direct role of PLD1 in adipogenic differentiation by regulating IRS-1 phosphorylation at serine 636/639 through DEPTOR displacement and mTOR activation.
Similar content being viewed by others

Introduction
Obesity is defined by excessive accumulation of white adipose tissue above the normal level of adipocyte differentiation owing to an energy imbalance. Dietary changes towards high protein and high fat intake have raised the prevalence of obesity over the last decade, increasing the risk of many disorders such as diabetes mellitus, hyperlipidemia, insulin resistance, cardiovascular disease, and cancer1,2,3. An understanding of the molecular mechanisms that regulate adipogenesis is required to reduce obesity and the accompanying susceptibility to many diseases. Adipocyte differentiation is a well-controlled process regulated by an elaborate network of transcription factors, including the CCAAT/enhancer-binding proteins C/EBPβ, C/EBPδ, C/EBPα and peroxisome proliferator-activated receptor γ (PPARγ)4,5. Expression of C/EBPβ and C/EBPδ is induced in preadipocytes during very early differentiation. Subsequently, these regulators activate PPARγ and C/EBPα, which upregulate each other and maintain their expression to govern the entire adipogenic process by activating additional transcription factors4.
The mammalian target of rapamycin (mTOR) pathway regulates many cellular and developmental processes by responding to growth factors and nutrients6. mTOR forms two distinct complexes: mTOR complex1 (mTORC1) and mTOR complex2 (mTORC2)7. mTORC1 controls translation and protein synthesis by phosphorylating ribosomal S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E binding protein 1 (4EBP1), whereas mTORC2 activates Akt, serum/glucocorticoid-regulated kinase (SGK), and protein kinase Cα (PKCα)8,9. Recently, a number of studies have shown that mTOR is involved in adipogenesis and lipid metabolism10,11,12,13. Inhibition of mTORC1 by either rapamycin treatment or an adipose-specific knockout of regulatory-associated protein of mTOR (also known as RPTOR or raptor, a major component of mTORC1), inhibits adipogenesis11,14. Conversely, activation of mTORC1 enhances adipogenesis by increasing PPARγ13, confirming a positive role for mTOR in adipogenesis15. Despite this, mTOR also maintains homeostasis of adipogenesis by suppressing the expression of PPARγ through insulin receptor substrate-1 (IRS-1)/Akt signaling16,17, suggesting an indispensable function for mTOR in adipogenesis.
Phospholipase D (PLD) hydrolyzes phosphatidylcholine (PC) to yield phosphatidic acid (PA) and choline18. PA is a critical regulator of mTOR signaling19. Of the several enzymes that are involved in PA biogenesis, PLD1 is the enzyme responsible for activating mTOR via either mitogen or amino acid stimulation19,20,21,22. PLD-produced PA is unique in that it contains fatty acid chains with one or two degrees of unsaturation23. Although generally PA is reported to maintain mTOR complexes in a steady-state condition24, PLD1-produced PA has been shown to specifically bind to the FRB domain of mTOR19 and displace an endogenous mTOR inhibitor, DEP domain-containing mTOR-interacting protein (DEPTOR), to activate mTORC125. In addition to the known role of PA in mTOR signaling, it has been suggested that PA regulates epidermal growth factor receptor (EGFR) trafficking from the membranes towards the nucleus and vice versa, and regulates EGFR expression in the nucleus, thus controlling EGF signaling26. PA is also involved in neuronal and endocrinal secretion by synaptic vesicle release. The biophysical properties of the PA molecule generate a negative curvature in the inner membrane leaflet of the plasma membrane and subsequently facilitate exocytosis27.
PLD has been implicated in a variety of cellular processes, such as the reorganization of actin cytoskeleton, membrane trafficking, secretion, receptor signaling, and differentiation18,28,29,30. Several studies using PLD-deficient mice have revealed physiological roles of PLD. Brain development, cognitive funtion31, and protection from thrombosis and ischemic stroke32 are all impaired in PLD-deficient mice. Mice lacking PLD1 have defects that result in tumor growth and metastasis33, and defects in macroautophagy34. Even though recent study has shown that PLD deficiency promotes adiposity by up-regulating appetite35, the role of PLD/PA in adipogenic differentiation remains unclear. Furthermore, it is not known whether PLD/PA is involved in the autonomous role of mTOR in adipogenesis, despite the fact that PLD and PA are well established as critical regulators of mTOR.
Here, we report that PLD1 plays a negative role in adipogenic differentiation. We observed that PLD1 and PA inhibit the initiation of adipogenesis through activation of mTORC1 via displacement of DEPTOR from mTORC1. We found that PA-activated mTORC1 preferentially phosphorylates IRS-1 at serine 636/639 and regulates the IRS-1/Akt signaling pathway. These results present PLD1 as a newly identified regulator of adipogenesis.
Results
PLD1 activity decreases during adipogenesis
We set out to test the activity and expression of PLD1 during adipogenic differentiation to examine its role in this process. 3T3-L1 preadipocytes were stimulated to differentiate by addition of differentiation medium 1 (DM1) with dexamethasone, IBMX, and insulin for two days, and then with differentiation medium 2 (DM2) for the following 4–6 days. The accumulation of lipid droplets was noticeable 3–4 days after induction, and adipocytes were fully differentiated after 7–8 days (Supplementary Fig. 1a). The mRNA levels of adipogenic markers (PPARγ, C/EBPα, adipocyte protein 2 (aP2)), and lipogenic markers (fatty acid synthase (FAS) and lipoprotein lipase (LPL)) were increased during differentiation (Supplementary Fig. 1b), as previously reported4,36,37. Measurements showed that total PLD activity during differentiation began to decrease on day 2 (Fig. 1a), and by day 4 PLD activity had dropped to almost 50% of the level seen in undifferentiated adipocytes (measured by 2 independent techniques; Fig. 1a and Supplementary Fig. 2a). Moreover, activity of the PLD isoform, PLD1, almost completely decreased by day 4, whereas the PLD2 activity was still present (Fig. 1b,c). This suggests that PLD1 is responsible for the majority of the decreased PLD activity observed during adipogenic differentiation. We observed that the expression of PLD1 and PLD2 increased slightly during adipocyte differentiation (Fig. 1d), implying that the decrease in PLD activity was not due to a decrease in PLD expression. Importantly, PLD activity decreased in white adipose tissue (WAT) of both high fat diet (HFD)-fed mice (Fig. 1e) and ob/ob mice (Fig. 1f), without any decrease in PLD1 mRNA expression (Supplementary Fig. 2b,c). These results imply that a reduction in PLD activity might be required for adipogenic differentiation.

PLD activity is decreased during 3T3-L1 adipogenic differentiation.
(a) 3T3-L1 cells were induced to differentiate for eight days. On day 0, 2, 4, 6 or 8 of differentiation, cells were lysed and a PLD assay was performed using a PLD assay kit. (b,c) (b) Recombinant PLD1 or PLD2 was transiently expressed. (c) Cells were differentiated for four days and recombinant in vivo PLD activities were measured using a transphosphatidylation assay as described in Materials and Methods. (d) Cells were treated as described in (a) and the lysates were subjected to western blotting. (e) White adipose tissue (WAT) was isolated from C57BL/6 J mice fed either a normal diet (ND) or a high-fat diet (HFD) for 12 weeks, lysed and subjected to PLD assay using a PLD assay kit (n = 4 per group). (f) WAT was isolated from either C57BL/6 J mice or ob/ob mice at age of 12 weeks and then subjected to PLD assay using a PLD assay kit (n = 9–10 per group). (g) Cells were differentiated in media under various conditions for four days and then in vivo PLD activities were measured with a transphosphatidylation assay. (h) 3T3-L1 cells were treated with 100 μM of 8-Br-cAMP and in vivo PLD activity was measured as in (g). (a–h) Data are representative of three to four independent experiments. Data are mean ± SD, with paired t-tests performed as indicated, *P < 0.05, **P < 0.01.
We proceeded to investigate if a component of the differentiation media was responsible for the decrease in PLD activity. We removed each component from the differentiation media individually, attempted to induce adipocyte differentiation with the resulting media iterations, and then measured PLD activity in each media environment. Removal of either IBMX or insulin increased PLD activity, whereas removal of dexamethasone had no effect on PLD activity (Fig. 1g). Although removal of any one component in the differentiation media decreased the overall rate of differentiation, the decreases did not correlate entirely with the changes observed in PLD activity (Supplementary Fig. 2d). Most notably, while removing insulin and IBMX together resulted in a level of PLD activity similar to that achieved by removing insulin or IBMX alone (last lane in Fig. 1g), the same simultaneous depletion caused a synergistic decrease in differentiation (last lane of Supplementary Fig. 2d). These results suggest that IBMX and insulin are responsible for the decrease in PLD activity, probably via the same signaling pathway, during 3T3-L1 differentiation. IBMX is a phosphodiesterase inhibitor that increases cAMP levels to strongly promote the initiation of adipogenic differentiation38. For this reason, we speculated that cAMP decreases PLD activity during adipogenesis; this was further supported by our observation that treating 3T3-L1 preadipocytes with cAMP decreased PLD activity (Fig. 1h), implying that PLD activity is affected by cAMP levels during adipogenesis.
PLD1 negatively regulates adipogenic differentiation
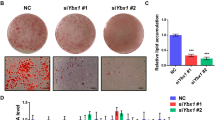
To further assess the role of PLD1 in adipogenesis, we performed a PLD1 knockdown using lentiviral shRNA delivery in 3T3-L1 cells. Two PLD1-targeting shRNA species decreased PLD1 protein levels by 94% and 75%, respectively (Fig. 2a). When these two PLD1-targeting shRNAs were expressed in 3T3-L1 cells, lipid droplet formation was significantly enhanced, as shown by Oil Red O staining (Fig. 2b). Quantification of Oil Red O staining revealed an increase of 3.07-fold and 1.88-fold in two PLD1 knockdown samples (Fig. 2b). Correspondingly, PLD1 deficiency resulted in an increase in mRNA levels of PPARγ, C/EBPα, aP2, and lipogenic genes such as LPL and FAS (Supplementary Fig. 3a), followed by elevated expression levels of PPARγ, C/EBPα, C/EBPβ, and aP2 protein upon differentiation (Fig. 2c). We then conducted similar experiments on mouse embryonic fibroblasts (MEFs) that were isolated from 13–15 day embryos of Pld1−/− mice. MEFs were induced to differentiate by adding rosiglitazone in DM1. Pld1−/− MEFs had more lipid droplets compared to Pld1+/+ MEFs, as shown by Oil Red O staining and its quantification (Fig. 2d). Pld1−/− MEFs also displayed clear increases in levels of the adipogenic differentiation markers PPARγ, C/EBPα, and aP2 over those seen in Pld1+/+ MEFs (Fig. 2e). Conversely, when we overexpressed PLD1 using adenoviral gene transfer in 3T3-L1 cells, lipid droplet formation on day 8 of differentiation was attenuated by up to 60% compared to control cells (Fig. 2f). Subsequently, mRNA levels of PPARγ, C/EBPα, aP2, and lipogenic genes such as FAS and LPL were decreased (Supplementary Fig. 3b), leading to reduced expression of PPARγ, C/EBPα, and aP2 protein (Fig. 2g). These results suggest that PLD1 is a critical negative regulator of adipogenesis.

PLD1 negatively regulates adipogenic differentiation.
(a) 3T3-L1 cells were infected with lentiviruses expressing two different PLD1 shRNAs or scrambled shRNA, and then selected by puromycin for 5 days. Cells were lysed and subjected to western blotting. Western blot intensities were analyzed using ImageJ. (b,c) Cells were treated as described in (a) and induced into differentiation for eight days. Cells were either (b) stained with Oil Red O and quantified for lipid contents, or (c) lysed and subjected to western blotting. (d,e) Pld1+/+ and Pld1−/− MEFs were differentiated for eight days and either (d) subjected to staining with Oil Red O and quantified for lipid contents, or (e) lysed and analyzed by western blotting. (f,g) Cells were induced to differentiate three days after the infection of adGFP or adPLD1, followed by either (f) Oil Red O staining and quantification for lipid content, or (g) lysed and analyzed by western blotting. (a–g) Images are representative of three to five experiments. Scale bars, 100 μm. All data shown are mean ± SD or blots representative of three to five independent experiments. Student t-tests were performed to compare the indicated pairs of data. *P < 0.05; **P < 0.01.
Inhibition of PLD1 activity induces adipogenesis
To address whether the enzymatic activity of PLD1 is required for the inhibitory effect of PLD1 during adipogenesis, we utilized VU0155069, a PLD1-specific inhibitor. Treatment of cells with VU0155069 upon induction of adipogenic differentiation increased lipid droplet formation by up to 36% compared to vehicle-treated cells (Fig. 3a). This is consistent with our observations in PLD1-deficient 3T3-L1 cells and Pld1−/− MEFs, specifically that PLD1 deficiency increased lipid accumulation. In addition, expression of adipogenic differentiation markers PPARγ, C/EBPα, and aP2 were elevated in VU0155069-treated cells (Fig. 3b and Supplementary Fig. 4), indicating that PLD1 negatively regulates adipogenesis. When VU0155069 was added for four days after day 4 of differentiation, adipogenic differentiation was not enhanced, as shown by lipid droplet formation (Fig. 3c) and expression of PPARγ, C/EBPα, and aP2 protein (Fig. 3d). These results suggest that PLD1 acts at the beginning of adipogenic differentiation, rather than during terminal differentiation or during maintenance of adipogenic characteristics in mature adipocytes.

Inhibition of PLD1 activity induces adipogenic differentiation.
(a,b) 3T3-L1 cells were differentiated in the presence of 5 μM VU0155069 at the initiation of differentiation for eight days. Cells were either (a) fixed and stained with Oil Red O and quantified for lipid content, or (b) lysed and analyzed by western blotting. (c,d) 3T3-L1 cells were induced to differentiate and 5 μM VU0155069 was added on day 4 of differentiation for four days. Cells were either (c) stained with Oil Red O and quantified for lipid content, or (d) lysed and analyzed by western blotting. (a–d) Images are representative of three to five experiments. Scale bars, 100 μm. All data shown are mean ± SD or blots representative of three to five independent experiments. Student t-tests were performed to compare the indicated pairs of data. **P < 0.01.
PA, a PLD1 product, inhibits adipogenesis
In order to determine whether phosphatidic acid (PA), a PLD1 product, is required for inhibition of adipogenic differentiation, we treated cells with a short chain PA (C8-PA) when differentiation was induced. We chose to use this PA species because it is not converted into active lysophosphatidic acid that could otherwise initiate signaling through membrane bound lysophosphatidic acid receptors39. As shown in Fig. 4a, PA treatment drastically decreased lipid formation, as shown by Oil Red O staining, and quantification revealed a reduction of up to 63% compared to control cells. Expression of PPARγ, C/EBPα, aP2 and lipogenic genes such as FAS and LPL was also reduced (Fig. 4b,c), which is consistent with the inhibitory effect of PLD1 expression on adipogenic differentiation. Interestingly, the augmented adipogenic differentiation in shPLD1-infected 3T3-L1 cells was reduced by either treatment with PA or by PLD1 expression via adPLD1 (adenovirus vector carrying the gene for PLD1), as shown by lipid content (Fig. 4d,f), mRNA levels of adipogenic and lipogenic markers such as PPARγ, C/EBPα, aP2, LPL and FAS (Supplementary Fig. 5a,b), and protein levels of PPARγ, C/EBPα, and aP2 (Fig. 4e,g). These results imply that PLD1 suppresses adipocyte differentiation through its enzymatic product, PA.

PA, the product of PLD1, inhibits adipogenesis.
(a) 3T3-L1 preadipocytes were differentiated with 300 μM C8-PA at the initiation of differentiation for eight days, followed by staining with Oil Red O and quantified for lipid content. (b,c) Cells were treated as described in (a), and then analyzed by either (b) quantitative RT-PCR or (c) western blotting. Mouse GAPDH was used to normalize gene expression. Cyclophilin A was used as an internal control. (d) 3T3-L1 cells were infected with either shscramble or shPLD1 and selected with puromycin for three days. Cells were induced to differentiate with or without 300 μM C8-PA. After eight days of differentiation, cells were fixed and stained with Oil Red O and quantified for lipid content. (e) Cells were treated as described in (d) and analyzed by western blotting. (f) Cells were infected with adenoviral PLD1 for three days in shPLD1-infected cells. Cells were then differentiated for eight days, followed by Oil Red O staining and quantification of lipid content. (g) Cells were treated as described in (f) and analyzed by western blotting. (a–g) Images are representative of three to five experiments. Scale bars, 100 μm. All data shown are mean ± SD or are blots representative of three to five independent experiments. Student t-tests were performed to compare the indicated pairs of data. *P < 0.05; **P < 0.01.
PA regulates adipogenic differentiation by displacing DEPTOR from mTORC1
Since PA decreased the expression of adipogenic genes during adipogenic differentiation, we considered it possible that PA directly regulates PPARγ activity—which itself coordinates expression of many genes responsible for the establishment of mature adipocytes40. To test this idea, we transfected 3T3-L1 adipocytes with a luciferase reporter that contained triple repeats of the DR1 sequence, a potent PPRE (PPARγ responsive element) and then induced differentiation. As shown in Fig. 5a, PA did not affect PPARγ activity. This suggests that PA regulates differentiation not by directly influencing PPARγ activity, but rather through a PA-induced signaling mechanism.

PA regulates adipogenic differentiation by displacing DEPTOR from mTORC1.
(a) 3T3-L1 preadipocytes were transfected with a PPRE luciferase reporter plasmid vector, induced to differentiate for 3 h, and then luciferase activity was measured. (b) 3T3-L1 cells were differentiated in the absence or presence of 300 μM C8-PA. Cell lysates and immunoprecipitates against raptor were analyzed by western blotting. (c,d) 3T3-L1 cells were infected by either shscramble or shDEPTOR and selected with puromycin for five days. After eight days of differentiation with or without 300 μM C8-PA, cells were subjected to either (c) Oil Red O staining and quantification of lipid content, or (d) western blotting. (a–d) All data shown are mean ± SD or are blots representative of three to five independent experiments. Student t-tests were performed to compare the indicated pairs of data. *P < 0.05.
PLD1-produced PA, which contains at least one unsaturated fatty acid, displaces DEPTOR, an endogenous mTORC1 inhibitor, from mTORC1 following mitogenic stimulation, resulting in mTORC1 activation25. DEPTOR expression is increased in WAT in an obese mouse model17. Moreover, DEPTOR enhances adipogenesis through Akt activation via mTORC1-induced phosphorylation of IRS-1 at serine 636/639, which is followed by PPARγ activation17. Therefore, in order to identify the mechanisms that underlie the inhibitory function of PA, we examined whether PA was able to displace DEPTOR from mTORC1 in 3T3-L1 cells. As shown in Supplementary Fig. 6, the level of DEPTOR in mTORC1 was significantly reduced in 3T3-L1 preadipocytes after 30 min treatment with PA. This is consistent with a previous report25. Notably, the level of DEPTOR in mTORC1 was increased during differentiation in 3T3-L1 cells (Fig. 5b; the second lane versus the third lane). PA treatment during differentiation reduced the level of DEPTOR in mTORC1 (Fig. 5b; the third lane versus last lane). In addition, DEPTOR knockdown by lentiviral delivery in 3T3-L1 cells drastically diminished lipid droplet formation as shown by Oil Red O staining and quantification (Supplementary Fig. 7a), and it reduced the expression of adipogenic differentiation markers such as PPARγ, C/EBPα, and aP2 (Supplementary Fig. 7b), confirming that DEPTOR is a positive regulator of adipogenesis (this is in agreement with a previous report)17. Since PA displaced DEPTOR from mTORC1 (Fig. 5b), we speculated that PA would have no further inhibitory effect on adipogenic differentiation in DEPTOR-knockdown 3T3-L1 cells, and we were able to confirm this experimentally. PA treatment did not further decrease adipogenic differentiation in DEPTOR-depleted cells, as shown by quantification of lipid content (Fig. 5c) and aP2 expression, a terminal differentiation marker (Fig. 5d). These results indicate that PA inhibits adipogenic differentiation via displacement of DEPTOR from mTORC1.
PA inhibits adipogenesis through the IRS-1/Akt pathway
To better understand the suppressive role of PA in adipogenesis, we determined the effect of PA on mTORC1 signaling in 3T3-L1 cells. PA has been reported to selectively regulate mTORC1 activity in vivo and in vitro25,39. As shown in Fig. 6a, treatment with PA for 30 minutes robustly increased IRS-1 phosphorylation at serine 636/639, without affecting phosphorylation at serine 1101. IRS1 phosphorylation at serine 307 was not detected in 3T3-L1 cells (data not shown). IRS1 phosphorylation at serine 636/639 is directly targeted by mTORC1 and induces IRS-1 degradation41. In addition, either PA treatment for eight days, or PLD1 overexpression by adenoviral PLD1 during 3T3-L1 adipogenic differentiation, increased S636/639-IRS-1 phosphorylation, which was followed by decreases in IRS-1 protein levels and by a decrease in Akt phosphorylation at serine 473 and threonine 308 (Fig. 6b,c). Notably, PA treatment did not affect any other classical mTORC1 downstream targets (S6K1, S6, 4EBP1) (Fig. 6b), indicating that, during adipogenesis, PLD1/PA does not regulate mTORC1 in the same manner as in other biological contexts. Conversely, depletion of PLD1 reduced S636/639-IRS-1 phosphorylation and increased Akt phosphorylation at S473 and T308 on day 8 of differentiation in either Pld1−/− MEFs (Fig. 6d) or shPLD1-infected cells (Fig. 6e). PA treatment restored S636/639-IRS-1 phosphorylation and decreased Akt phosphorylation in PLD1 knockdown 3T3-L1 cells (Fig. 6e; the second lane versus the third lane). However, PA treatment did not cause a further increase in S636/639-IRS-1 phosphorylation in DEPTOR-depleted cells, in which S636/639-IRS-1 phosphorylation was already increased (Fig. 6f). These results indicate that PA induces IRS-1 phosphorylation via displacement of DEPTOR from mTORC1.

PA inhibits adipogenesis through the IRS-1-Akt axis.
(a) 3T3-L1 preadipocytes were serum-starved overnight, then stimulated with 300 μM C8-PA for 30 min. Cell lysates were analyzed by western blotting. (b) 3T3-L1 cells were induced to differentiate either with or without 300 μM C8-PA for eight days, then lysed and subjected to western blotting. (c) 3T3-L1 cells were infected with either adGFP or adPLD1, induced to differentiate for eight days, and then analyzed by western blotting. (d) Pld1+/+ and Pld1−/− MEFs were differentiated for eight days, lysed and analyzed by western blotting. (e) Cells were infected with either shscramble or shPLD1 #1, selected with puromycin for five days, then induced to differentiate either with or without 300 μM PA for eight days, followed by analysis by western blotting. (f) 3T3-L1 cells were infected with either shscramble or shDEPTOR and treated either with or without 300 μM PA for 30 min, then lysed and analyzed by western blotting. (a–f) All data shown are blots representative of three to five independent experiments.
Discussion
Adipogenic differentiation is a cellular and developmental process that is important for metabolic homeostasis. In the present study, we provide evidence that PLD1 regulates adipogenic fat accumulation. In the early stages of adipogenesis, a decrease in PLD1 activity elevates adipogenic differentiation by up-regulating DEPTOR levels in mTORC1, and consequently, down-regulating mTORC1 activity. In describing this regulatory pathway, we identified PLD1 as a negative regulator of adipogenesis.
PLD activity was previously reported to be indispensable for lipid droplet formation by enhancing ERK2-induced dynein phosphorylation42,43, pointing to a role for PA in lipid metabolism. In order to maintain phospholipid flux, PA is converted to diacylglycerol (DAG), which is further metabolized into either triglycerides (TG) or phosphatidylcholine (PC)44, followed by enhanced lipid accumulation. However, in the present study, either PLD overexpression or PA treatment resulted in decreased rates of adipogenic differentiation, suggesting that PLD1 does not metabolize lipids and form lipid droplets during adipogenic differentiation. We found that PLD activity was reduced by both IBMX and insulin (Fig. 1g). IBMX inhibits phosphodiesterases, resulting in an increase in cellular cAMP5. Upon adipogenic differentiation, high cAMP levels lead to activation of C/EBPα and PPARγ, and also drive aP2 expression45,46. Previous reports showed that cAMP or cAMP-increasing agents inhibit PLD activity in vitro and in vivo47,48,49. In agreement with these reports, we observed that cAMP decreased PLD activity (Fig. 1h), suggesting that PLD is a downstream target of cAMP in adipogenesis. Unlike cAMP, insulin was reported to activate PLD activity in 3T3-L1 cells, which peaked within the first 2 minutes and then returned to basal levels50. It is therefore plausible that the decreased PLD activity we observed in the present study four days after differentiation may not be a direct effect of insulin on PLD activity. Supporting this idea, cotreatment with IBMX and insulin decreased PLD activity to an extent similar to treatment with either IBMX alone or insulin alone, suggesting that IBMX and insulin regulate PLD activity through the same pathway, possibly regulating cAMP levels. The mechanism by which insulin regulates PLD activity during 3T3-L1 differentiation requires further investigation.
PLD activity was decreased in both HFD-induced obese mice (Fig. 1e) and ob/ob mice (Fig. 1f), indicating that reduction of PLD activity correlates with obesity in physiological contexts. Recently, the role of PLD1 and PLD2 has been implicated in protecting against excessive weight gain35. Mice lacking either PLD1 or PLD2 consumed more food compared to control mice; this was accompanied by the expression of neuropeptides controlling food intake in the hypothalamus. Subsequently, mice lacking either PLD1 or PLD2 exhibited elevated body weight and adipose tissue content. Although this finding supports that PLD1 is a negative regulator of adipogenesis, it is most unlikely that PLD deficiency elicits obesity simply due to a defect in adipose tissue. Studies of adipose-tissue-specific PLD1 knockout mice are needed to confirm our observations in vivo, and to assess any adipose tissue-specific contribution to obesity.
PA is known to prompt signaling events, such as mTOR signaling19,25. The role of mTOR signaling in adipogenesis is complex, involving a balance of positive and negative regulatory functions. Through the present study, we clarified one piece of this puzzle by demonstrating that the upstream regulators, PLD1 and PA, preferentially regulate mTORC1’s negative function in adipogenic differentiation. During 3T3-L1 differentiation, PLD1/PA activates mTORC1 by displacing DEPTOR and phosphorylating S636/639-IRS-1, which is a direct target of mTORC1 and promotes IRS-1 degradation and inhibition of adipogenesis. Notably, PA did not augment the phosphorylation of S6K1/S6 and 4EBP1—two well-known downstream effectors of mTORC1—during adipogenesis. These results suggest that mTORC1 activation by DEPTOR displacement in differentiating adipocytes selectively induces the negative feedback on IRS-1, without affecting mTORC1 regulation of other substrates. In contexts where mTORC1 and, therefore, S6K1 are completely inhibited, there is a subsequent decrease of S6K1-facilitated adipogenic gene expression. This precludes any significant positive contribution of the IRS-1/Akt pathway to adipogenesis, despite the fact that that pathway is no longer inhibited by mTORC116,17. Conversely, partial inhibition of mTOR, which relieves the direct phosphorylation and negative regulation of IRS-1 and subsequently increases Akt activity while preserving S6K1 signaling, increases adipogenesis by way of enhanced lipid accumulation and adipogenic marker expression16. In the present study, we have identified a third context wherein, in differentiating adipocytes with high levels of DEPTOR, PA preferentially stimulates phosphorylation of S636/639-IRS-1, while leaving T389-S6K1 phosphorylation levels unchanged. It appears that these high DEPTOR expression levels limit the extent of mTORC1 activation by PA, resulting in mild mTORC1 activation that is specific to, and directly responsible for, downregulation of the IRS-1/Akt pathway.
PA has been reported to regulate the membrane association of lipin1β via binding to the polybasic motif of lipin1β, a protein which has dual functions as a factor in intracellular lipid metabolism and in transcriptional control of adipogenesis44. Maintenance of the distribution of lipin1β at both the intracellular membrane and within the nucleus is critically important to the biological activity of lipin1β44. Given the role of PA in maintaining lipin1β distribution, both PA deficiency and PA increase were expected to inhibit adipogenesis due to a disruption in the balance of lipin1β localization. However, the current study shows that PA deficiency increases adipocyte differentiation while PA treatment decreases it, indicating that the role of PA in adipogenesis is not related to regulation of the balance of lipin1β between the nucleus and cytoplasm.
PLD2, an isoform of PLD, produces cyclic phosphatidic acid (CPA) from lysophosphatidylcholine. CPA directly binds to PPARγ with nanomolar affinity and stabilizes the binding of a corepressor, silencing mediator of retinoid and thyroid hormone receptors (SMRT), to PPARγ — resulting in inhibition of PPARγ-related gene expression51,52. CPA also inhibits phosphodiesterase 3B (PDE3B) expression and subsequently increases cAMP, leading to increased lipolysis in 3T3-L1 cells and an inhibition of adipogenesis53. Even though both PLD1 and PLD2 hydrolyze PC to produce PA, PLD1 is not involved in insulin-stimulated CPA production52. Moreover, our results show that PA does not directly modulate PPARγ activity (Fig. 5a), suggesting that PLD1-induced inhibition of adipogenesis is not the result of a direct effect of PA on PPARγ activity.
Notably, expression of PLD1 and PLD2 mildly increased during adipogenic differentiation, despite the fact that PLD functions as a negative regulator. It was reported that the expression levels of negative regulators increased during adipogenic differentiation54 as well as during other forms of cellular differentiation, such as myogenesis55,56. Since PLD/PA controls adipogenesis through several different pathways (e.g., lipin1β localization, PPARγ activity through PLD2-prduced CPA, mTOR signaling through DEPTOR), precise regulation of PLD is required for the fine control of adipogenic differentiation. The increased expression of PLD might introduce a necessary negative feedback loop that prevents excessive differentiation.
In conclusion, we have shown that PLD1 inhibits adipogenesis via specific induction of a negative role for mTORC1. These findings improve our insights into the regulation of PLD1 in mTOR signaling and increase our understanding of the molecular mechanisms that underlie adipogenic differentiation. Furthermore, these new molecular discoveries may prompt the development of PLD1-specific activation methods as a therapeutic strategy for treating obesity.
Materials and Methods
Cell lysis, immunoprecipitation, western blotting, Oil Red O staining and lipid content assay, and quantitative RT-PCR were all performed following standard procedures or as previously reported, with detailed information described in the Supplementary information.
Antibodies and other reagents
Antibodies against the following proteins were obtained from the following sources: alpha-tubulin (ab11304) from Abcam; DEPTOR (NBP149674) from Novus Biologicals; raptor (for IP, A300-553A) from Bethyl laboratories; PLD2 (sc-25513), PPARγ (sc-7196), C/EBPα (sc-61) and aP2 (sc-271529) from Santa Cruz Biotechnology; C/EBPβ (#3087), C/EBPδ (#2318), IRS-1 (#2382), pS636/639-IRS-1 (#2388), pS1101-IRS-1 (#2385), mTOR (#2972), raptor (#2280), Akt (#9272), pS473-Akt (#4051), pT308-Akt (#9275), S6K1 (#9202), pT389-S6K1 (#9234), S6 (#2217), pSer235/236-S6 (#4856), 4EBP1 (#9644), and pT37/46-4EBP1 (#2855) from Cell Signaling Technology. The PLD1 antibody was provided by Dr. Jie Chen from the University of Illinois at Urbana-Champaign16. All secondary antibodies were from Jackson ImmunoResearch Laboratories Inc. [9,10-3H(N)]-oleic acid was from PerkinElmer. C8-PA (830842C) was from Avanti. All other reagents were from Sigma-Aldrich.
Cell culture
3T3-L1 preadipocytes were grown at 37 °C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose and 10% bovine calf serum (M1). For 3T3-L1 preadipocyte differentiation, 2 days after the cells were confluent in M1 (day 0), the cells were induced to differentiate in M2 (DMEM containing 10% fetal bovine serum [FBS], 1.0 μM dexamethasone, 0.5 mM IBMX, and 1.5 μg/mL insulin). After 48 hours, M2 medium was replaced by M3 (DMEM containing 10% FBS and 1.5 μg/mL insulin), and cells were fed every 2 days with M3. At the time points indicated, cells were harvested for RT-PCR, western blotting, or Oil Red O staining. Pld1−/− mice were generated and characterized as described34. To prepare Pld1+/+ and Pld1−/− MEFs, 13–15 day post-coital mouse embryos were minced and digested with trypsin. The cells were collected and cultured in DMEM with 4.5 mg/L glucose, 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. MEFs were induced to differentiate by adding rosiglitazone in DM1.
Animals
For diet-induced obese mice, 7-week-old C57BL/6 J mice were obtained from the Jackson Laboratory. After adaptation for one week, mice were provided with either a HFD (60% fat) or regular chow diet (5.4% fat) for 12 weeks. C57BL/6 J mice and ob/ob mice were obtained from Daehan Bio Link (Daejeon, Korea) and Japan SLC (Hamamatsu, Japan), respectively. Food and water were provided ad libitum, and the mice were kept on a 12 h light, 12 h dark cycle. Male mice were used for all studies. All experimental protocols for animals, maintenance and care, were conducted in accordance with Gachon University Animal Care guidelines. All animal procedures were approved by the Gachon University of Medicine and the Institutional Animal Care and Use Committee (IACUC).
PLD assays
In vivo PLD activity was determined by PLD-catalyzed transphosphatidylation as previously described20. Briefly, 3T3-L1 cells were metabolically labeled with 3H-oleic acid for 18 h. After pretreatment with 0.3% 1-butanol for 30 min, cells were lysed and lipids were extracted according to the method of Bligh and Dyer57 and analyzed by TLC. Alternatively, a Phospholipase D Assay Kit (Sigma Aldrich) was also used, as indicated in the figure legends, to measure PLD activity according to the manufacturer’s instructions58,59,60,61. To calculate recombinant PLD1 or PLD2 activity specifically, the PLD activity in cells transfected with empty vector was subtracted from the activity in PLD1 or PLD2-transfected cells under the same conditions20,62. Cells were transfected with HA-PLD120 and HA-PLD220 using Amaxa Nucleofector technology (Amaxa, Cologne, Germany) according to manufacturers’ protocol.
Lentivirus-mediated shRNA and adenovirus-mediated gene expression
Lentiviral shRNA plasmids were used in the pLKO.1puro vector and lentivirus, and were packaged into 293T cells as previously described30. Mouse PLD1 shRNA#1 (TRCN0000076820), mouse PLD1 shRNA#2 (TRCN0000076822), negative control shRNA (scrambled hairpin sequence), mouse DEPTOR shRNA #1 (Addgene 21337), and mouse DEPTOR shRNA #2 (Addgene 21338) were used as previously described30,63. After viral infection, selection of infected cells with 4 μg/mL puromycin was performed for 3–5 days before differentiation was induced. 3T3-L1 cells were incubated with recombinant adenoviruses bearing human PLD1 cDNA or the GFP gene for 24 h and induced to differentiate eight days after infection. Adenoviral PLD1 and adenoviral GFP were used as previously reported64.
Luciferase assays
Cells were transfected with the PPARγ responsive element (PPRE) luciferase reporter (3x DR1) plasmid vector14 using the TransIT-LT1 transfection reagent (Mirus) following manufacturers’ recommendations. Cells were induced into differentiation for 3 h and lysed in Passive Lysis buffer (Promega). Luciferase assays were performed using a Luciferase Assay System kit (Promega) following the manufacturer’s protocol.
Statistical analysis
All numerical data are presented as mean ± standard deviation (SD). Where necessary, statistical significance was determined by performing one-sample t-tests. P-values <0.05 were considered statistically significant.
Additional Information
How to cite this article: Song, H.-I. and Yoon, M.-S. PLD1 regulates adipogenic differentiation through mTOR - IRS-1 phosphorylation at serine 636/639. Sci. Rep. 6, 36968; doi: 10.1038/srep36968 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Wang, Y., Rimm, E. B., Stampfer, M. J., Willett, W. C. & Hu, F. B. Comparison of abdominal adiposity and overall obesity in predicting risk of type 2 diabetes among men. Am J Clin Nutr 81, 555–563 (2005).
Pischon, T. et al. General and abdominal adiposity and risk of death in Europe. N Engl J Med 359, 2105–2120 (2008).
Flegal, K. M., Williamson, D. F. & Graubard, B. I. Obesity and cancer. N Engl J Med 349, 502–504, author reply 502–504 (2003).
Rosen, E. D., Walkey, C. J., Puigserver, P. & Spiegelman, B. M. Transcriptional regulation of adipogenesis. Genes Dev 14, 1293–1307 (2000).
Farmer, S. R. Transcriptional control of adipocyte formation. Cell Metab 4, 263–273 (2006).
Wullschleger, S., Loewith, R. & Hall, M. N. TOR signaling in growth and metabolism. Cell 124, 471–484 (2006).
Zoncu, R., Efeyan, A. & Sabatini, D. M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12, 21–35 (2011).
Lamming, D. W. & Sabatini, D. M. A Central role for mTOR in lipid homeostasis. Cell Metab 18, 465–469 (2013).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Cho, H. J., Park, J., Lee, H. W., Lee, Y. S. & Kim, J. B. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem Biophys Res Commun 321, 942–948 (2004).
Polak, P. et al. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8, 399–410 (2008).
Yu, W. et al. Critical role of phosphoinositide 3-kinase cascade in adipogenesis of human mesenchymal stem cells. Mol Cell Biochem 310, 11–18 (2008).
Zhang, H. H. et al. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One 4, e6189 (2009).
Kim, J. E. & Chen, J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 53, 2748–2756 (2004).
Yi, S. A. et al. S6K1 Phosphorylation of H2B Mediates EZH2 Trimethylation of H3: A Determinant of Early Adipogenesis. Mol Cell 62, 443–452 (2016).
Yoon, M. S., Zhang, C., Sun, Y., Schoenherr, C. J. & Chen, J. Mechanistic target of rapamycin controls homeostasis of adipogenesis. J Lipid Res 54, 2166–2173 (2013).
Laplante, M. et al. DEPTOR cell-autonomously promotes adipogenesis, and its expression is associated with obesity. Cell Metab 16, 202–212 (2012).
McDermott, M., Wakelam, M. J. & Morris, A. J. Phospholipase D. Biochemistry and cell biology = Biochimie et biologie cellulaire 82, 225–253 (2004).
Fang, Y., Vilella-Bach, M., Bachmann, R., Flanigan, A. & Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294, 1942–1945 (2001).
Yoon, M. S., Du, G., Backer, J. M., Frohman, M. A. & Chen, J. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. The Journal of cell biology 195, 435–447 (2011).
Xu, L. et al. Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1). J Biol Chem 286, 25477–25486 (2011).
Fang, Y. et al. PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Curr Biol 13, 2037–2044 (2003).
Pettitt, T. R., McDermott, M., Saqib, K. M., Shimwell, N. & Wakelam, M. J. Phospholipase D1b and D2a generate structurally identical phosphatidic acid species in mammalian cells. Biochem J 360, 707–715 (2001).
Toschi, A. et al. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol 29, 1411–1420 (2009).
Yoon, M. S. et al. Rapid Mitogenic Regulation of the mTORC1 Inhibitor, DEPTOR, by Phosphatidic Acid. Mol Cell 58, 549–556 (2015).
Henkels, K. M. et al. A Phosphatidic Acid (PA) conveyor system of continuous intracellular transport from cell membrane to nucleus maintains EGF receptor homeostasis. Oncotarget (2016).
Zeniou-Meyer, M. et al. Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J Biol Chem 282, 21746–21757 (2007).
Huang, P. & Frohman, M. A. The potential for phospholipase D as a new therapeutic target. Expert Opin Ther Targets 11, 707–716 (2007).
Yoon, M. S. et al. Phospholipase D1 as a key enzyme for decidualization in human endometrial stromal cells. Biol Reprod 76, 250–258 (2007).
Yoon, M. S. & Chen, J. PLD regulates myoblast differentiation through the mTOR-IGF2 pathway. J Cell Sci 121, 282–289 (2008).
Burkhardt, U. et al. Impaired brain development and reduced cognitive function in phospholipase D-deficient mice. Neuroscience letters 572, 48–52 (2014).
Thielmann, I. et al. Redundant functions of phospholipases D1 and D2 in platelet alpha-granule release. Journal of thrombosis and haemostasis: JTH 10, 2361–2372 (2012).
Chen, Q. et al. Key roles for the lipid signaling enzyme phospholipase d1 in the tumor microenvironment during tumor angiogenesis and metastasis. Science signaling 5, ra79 (2012).
Dall’Armi, C. et al. The phospholipase D1 pathway modulates macroautophagy. Nature communications 1, 142 (2010).
Trujillo Viera, J., El-Merahbi, R., Nieswandt, B., Stegner, D. & Sumara, G. Phospholipases D1 and D2 Suppress Appetite and Protect against Overweight. PLoS One 11, e0157607 (2016).
Kim, J. B. & Spiegelman, B. M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev 10, 1096–1107 (1996).
Rosen, E. D. et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev 16, 22–26 (2002).
Petersen, R. K. et al. Cyclic AMP (cAMP)-Mediated Stimulation of Adipocyte Differentiation Requires the Synergistic Action of Epac- and cAMP-Dependent Protein Kinase-Dependent Processes. Molecular and Cellular Biology 28, 3804–3816 (2008).
Yoon, M. S., Sun, Y., Arauz, E., Jiang, Y. & Chen, J. Phosphatidic acid activates mammalian target of rapamycin complex 1 (mTORC1) kinase by displacing FK506 binding protein 38 (FKBP38) and exerting an allosteric effect. J Biol Chem 286, 29568–29574 (2011).
Farmer, S. R. Regulation of PPAR[gamma] activity during adipogenesis. Int J Obes Relat Metab Disord 29, S13–S16 (2005).
Hiratani, K. et al. Roles of mTOR and JNK in serine phosphorylation, translocation, and degradation of IRS-1. Biochem Biophys Res Commun 335, 836–842 (2005).
Marchesan, D. et al. A phospholipase D-dependent process forms lipid droplets containing caveolin, adipocyte differentiation-related protein, and vimentin in a cell-free system. J Biol Chem 278, 27293–27300 (2003).
Andersson, L. et al. PLD1 and ERK2 regulate cytosolic lipid droplet formation. J Cell Sci 119, 2246–2257 (2006).
Ren, H. et al. A phosphatidic acid binding/nuclear localization motif determines lipin1 function in lipid metabolism and adipogenesis. Mol Biol Cell 21, 3171–3181 (2010).
Sun, L., Nicholson, A. C., Hajjar, D. P., Gotto, A. M. & Han, J. Adipogenic differentiating agents regulate expression of fatty acid binding protein and CD36 in the J744 macrophage cell line. Journal of Lipid Research 44, 1877–1886 (2003).
Yang, V. W., Christy, R. J., Cook, J. S., Kelly, T. J. & Lane, M. D. Mechanism of regulation of the 422(aP2) gene by cAMP during preadipocyte differentiation. Proceedings of the National Academy of Sciences 86, 3629–3633 (1989).
Agwu, D. E., McCall, C. E. & McPhail, L. C. Regulation of phospholipase D-induced hydrolysis of choline-containing phosphoglycerides by cyclic AMP in human neutrophils. J Immunol 146, 3895–3903 (1991).
Kwak, J. Y. & Uhlinger, D. J. Downregulation of phospholipase D by protein kinase A in a cell-free system of human neutrophils. Biochem Biophys Res Commun 267, 305–310 (2000).
Tyagi, S. R., Olson, S. C., Burnham, D. N. & Lambeth, J. D. Cyclic AMP-elevating agents block chemoattractant activation of diradylglycerol generation by inhibiting phospholipase D activation. J Biol Chem 266, 3498–3504 (1991).
Huang, P., Altshuller, Y. M., Hou, J. C., Pessin, J. E. & Frohman, M. A. Insulin-stimulated Plasma Membrane Fusion of Glut4 Glucose Transporter-containing Vesicles Is Regulated by Phospholipase D1. Mol Biol Cell 16, 2614–2623 (2005).
Oishi-Tanaka, Y. & Glass, C. K. A new role for cyclic phosphatidic acid as a PPARgamma antagonist. Cell Metab 12, 207–208 (2010).
Tsukahara, T. et al. Phospholipase D2-dependent inhibition of the nuclear hormone receptor PPARgamma by cyclic phosphatidic acid. Mol Cell 39, 421–432 (2010).
Tsukahara, T., Hanazawa, S. & Murakami-Murofushi, K. Cyclic phosphatidic acid influences the expression and regulation of cyclic nucleotide phosphodiesterase 3B and lipolysis in 3T3-L1 cells. Biochem Biophys Res Commun 404, 109–114 (2011).
Kim, J. H. et al. AIMP1 negatively regulates adipogenesis by inhibiting PPARγ. Journal of Cell Science 127, 4483–4493 (2014).
Liu, G. H. et al. Regulation of myoblast differentiation by the nuclear envelope protein NET39. Mol Cell Biol 29, 5800–5812 (2009).
Lee, C. S. et al. TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death Differ 17, 1254–1265 (2010).
Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37, 911–917 (1959).
Chaim, O. M. et al. Phospholipase-D activity and inflammatory response induced by brown spider dermonecrotic toxin: endothelial cell membrane phospholipids as targets for toxicity. Biochim Biophys Acta 1811, 84–96 (2011).
Liu, Y. et al. The role of phospholipase D in modulating the MTOR signaling pathway in polycystic kidney disease. PLoS One 8, e73173 (2013).
Elvers, M. et al. Impaired alpha(IIb)beta(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Science signaling 3, ra1 (2010).
Momboisse, F. et al. betaPIX-activated Rac1 stimulates the activation of phospholipase D, which is associated with exocytosis in neuroendocrine cells. J Cell Sci 122, 798–806 (2009).
Colley, W. C. et al. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol 7, 191–201 (1997).
Peterson, T. R. et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886 (2009).
Kang, D. W. et al. Targeting phospholipase D1 attenuates intestinal tumorigenesis by controlling beta-catenin signaling in cancer-initiating cells. J Exp Med 212, 1219–1237 (2015).
Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2015R1D1A1A01058313), and the Gachon University Gil Medical Center (FRD2015-15, FRD2014-12-02).
Author information
Authors and Affiliations
Contributions
H.-I.S. and M.-S.Y. designed and performed all research, analyzed all data, and wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Song, HI., Yoon, MS. PLD1 regulates adipogenic differentiation through mTOR - IRS-1 phosphorylation at serine 636/639. Sci Rep 6, 36968 (2016). https://doi.org/10.1038/srep36968
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36968
This article is cited by
-
Transcription factor EB regulates phosphatidylinositol-3-phosphate levels that control lysosome positioning in the bladder cancer model
Communications Biology (2023)
-
Liver-derived cell lines from cavefish Astyanax mexicanus as an in vitro model for studying metabolic adaptation
Scientific Reports (2022)
-
Role of pirfenidone in TGF-β pathways and other inflammatory pathways in acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection: a theoretical perspective
Pharmacological Reports (2021)
-
Saturated fatty acids induce insulin resistance in podocytes through inhibition of IRS1 via activation of both IKKβ and mTORC1
Scientific Reports (2020)
-
Identification of somatic alterations in lipoma using whole exome sequencing
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
