
Abstract
Mutations in the ganglioside-induced differentiation associated protein 1 (GDAP1) cause severe peripheral motor and sensory neuropathies called Charcot-Marie-Tooth disease. GDAP1 expression induces fission of mitochondria and peroxisomes by a currently elusive mechanism, while disease causing mutations in GDAP1 impede the protein’s role in mitochondrial dynamics. In silico analysis reveals sequence similarities of GDAP1 to glutathione S-transferases (GSTs). However, a proof of GST activity and its possible impact on membrane dynamics are lacking to date. Using recombinant protein, we demonstrate for the first time theta-class-like GST activity for GDAP1, and it’s activity being regulated by the C-terminal hydrophobic domain 1 (HD1) of GDAP1 in an autoinhibitory manner. Moreover, we show that the HD1 amphipathic pattern is required to induce membrane dynamics by GDAP1. As both, fission and GST activities of GDAP1, are critically dependent on HD1, we propose that GDAP1 undergoes a molecular switch, turning from a pro-fission active to an auto-inhibited inactive conformation.
Similar content being viewed by others


Introduction
Ganglioside-induced differentiation associated protein 1 (GDAP1) is an integral, tail-anchored protein of the mitochondrial outer membrane (MOM) and the peroxisomal membrane1,2,3. GDAP1 is predominantly expressed in neural cells where it mediates mitochondrial and peroxisomal fragmentation dependent on ubiquitously expressed fission factors (Fis1, Drp1, and Mff)1,2,3,4,5. Mutations in GDAP1 are associated with the hereditary motor and sensory neuropathy Charcot-Marie-Tooth (CMT) disease6,7,8. In vitro studies revealed that recessively inherited mutant forms of GDAP1 (rmGDAP1) exhibit reduced fission-promoting activity, whereas dominantly inherited mutant forms (dmGDAP1) interfere with mitochondrial fusion4,9. Furthermore, while GDAP1 expression is protective in glutamate-induced toxicity, this protection is reduced in rmGDAP1s, dependent on their residual fission-capacity10.
In silico analyses predict that GDAP1 is a glutathione S-transferase (GST)11,12,13. GSTs are multifunctional proteins involved in cellular detoxification or regulating cellular glutathione (GSH) levels, hormone biosynthesis, and intracellular signaling14,15. These enzymes are classified according to their protein sequence and structure, dimerization motif, features of the catalytic active centre, and substrate specificity14,15,16. GDAP1 is an integral membrane protein and contains in addition to its transmembrane domain (TMD) a further C-terminal hydrophobic domain (HD1). Two domains in GDAP1 (GST-N and GST-C) show similarities to cytosolic zeta-, omega-, or theta-class GSTs11,13. Between its putative GST-N and GST-C domains, GDAP1 contains an extended interdomain linker which may adopt two additional alpha-helices13. These features are also present in the GDAP1-paralogue GDAP1like1, possibly constituting a new class of GST proteins13. In response to changes in the cellular redox state, the cytosolic GDAP1like1 translocates to mitochondria, integrates into the MOM and causes mitochondrial fission17. This translocation and integration of GDAP1like1 into the MOM is specifically caused by an increase in the concentration of the oxidized form of glutathione in vitro and in vivo where it is capable of substituting for the loss of GDAP1 in the central nervous system of GDAP1-deficient mice17. Interestingly, mitochondrial translocation from the cytosol under oxidative stress conditions has previously been described also for other GSTs such as GSTA4–4 and GSTPi18,19,20,21. However, in spite of considerable effort, attempts to demonstrate GST activity of GDAP1 and the functional consequences have not met success to date5,11,12,13.
We show here that highly purified recombinant human GDAP1 is indeed a GST enzyme, and we demonstrate specific GSH-conjugating activity in vitro. We discovered that GST activity is regulated by the hydrophobic domain 1 (HD1), which exerts an autoinhibitory function. HD1 could adopt an amphipathic pattern and this amphipathic pattern is necessary to induce remodelling of organelles-mimicking liposomes by Gdap1. We propose a model of action with two different, interconvertible conformations of GDAP1: a GST active state with an exposed HD1 mediating mitochondrial and peroxisomal fission, and a GST-inactive state caused by an autoinhibitory binding mode of HD1.
Results
GDAP1 forms dimers
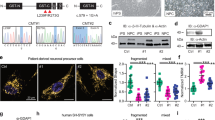
GST enzymes critically depend on dimerization in order to be catalytically active14. To test the dimerization capacity of GDAP1, we transiently co-expressed human and mouse isoforms of full-length and untagged GDAP1 or disease-causing mutant forms of GDAP1 (Fig. 1A) in HEK-293T cells and performed co-immunoprecipitation (co-IP) experiments using human-isoform-specific anti-GDAP1 antibodies. IPs of human GDAP1 co-precipitated mouse Gdap1 (Fig. 1B). In addition, all tested rmGDAP1 (R120Q, R310Q) and dmGDAP1 (R120W, Q218E) were also able to form homodimers (Fig. 1B). Consistently, human GDAP1 was co-precipitated with the murine specific Gdap1 antibody (see Supplementary Fig. S1A), confirming that GDAP1 wildtype or disease mutants can dimerize.

GDAP1 forms homodimers.
(A) Schematic representation of the domain structure of GDAP1 (conserved GST domains: GST-N and GST-C; hydrophobic domain 1: HD1, transmembrane domain: TMD). The localization of disease-associated missense mutations used in this study and GDAP1 truncations lacking the TMD (GDAP1 T318X) or TMD and HD1 (GDAP1 T288X) are illustrated. Human and mouse-specific antisera were generated against species-specific epitopes of the N-terminal region (epitopes). (B,C) Immunoprecipitation performed with the human-specific anti-GDAP1 antibody co-precipitates the corresponding mouse isoforms from co-transfected HEK-293T cells. Without human GDAP1 co-expression, the murine GDAP1 is not precipitated. *Signal of IgG heavy chains. (D) S200 elution profiles and a Coomassie blue stained SDS-gel of the purified proteins. Peak fractions of T318X-6xHis were pooled separately and re-chromatographed under identical conditions (E) consistently resulting in 2 peaks.
We next tested whether the C-terminal hydrophobic domains of GDAP1 are dispensable for dimerization. GDAP1 lacking the TMD (GDAP1 T318X) or lacking the TMD and HD1 (GDAP1 T288X) were co-expressed as human and murine truncated proteins in HEK-293T cells (Fig. 1A). Co-immunoprecipitation revealed that both truncated forms homo-dimerize (Fig. 1C, see Supplementary Fig. S1B). These data are consistent with previous experiments using truncated or tagged GDAP1 constructs11,22. We conclude that GDAP1 T288X and GDAP1 T318X are capable of dimerization, which is a pre-requisite for catalytic activity.
GDAP1 is catalytically active and mediates glutathione-conjugation in vitro
Recombinant, full-length or truncated GDAP1 expressed in bacteria is not soluble (data not shown), consistent with previous reports11. We therefore tested eukaryotic expression using MultiBac, a baculovirus-based insect cell protein expression system23. We expressed two soluble His-tagged GDAP1 variants (GDAP1 T288X-6xHis and GDAP1 T318X-6xHis) in large amounts. The proteins were purified to homogeneity by metal affinity chromatography followed by size-exclusion chromatography (SEC) (Fig. 1D). Elution profiles showed two peaks, indicating that both GDAP1 variants exist in a monomeric and a dimeric form, validating our co-immunoprecipitation experiments (Fig. 1E). Re-chromatographing either the monomeric or dimeric peak fractions by SEC resulted again in two peaks, consistent with a dynamic monomer-dimer equilibrium (Fig. 1E).
We assayed GST activity with the highly purified GDAP1 proteins using different model substrates. Our experiments revealed GSH-conjugating activity for GDAP1 T288X to ethacrynic acid, p-Nitrobenzylchloride, and 1,2-Epoxy-3-(4-nitrophenoxy)propane, but not to the classic GST-substrate 1-Chloro-2,4-dinitrobenzene (Table 1). In addition, both GDAP1 variants failed to bind GSH-sepharose (not shown). This activity profile is characteristic for theta-class GSTs16. Our results thus unequivocally demonstrate that GDAP1 has theta-class-like GST activity in vitro11,13,16.
HD1 regulates GDAP1 GST- activity and mediates membrane curvature in vitro
Strikingly, in contrast to GDAP1 T288X-6xHis, the longer construct GDAP1 T318X-6xHis containing the C-terminal HD1, did not display detectable GST activity with any of the substrates tested (Table 1). These results suggest a regulatory function of HD1 acting as an autoinhibitory domain, possibly by locking the protein in an inactive conformation. Interestingly, HD1 is essential to execute GDAP1-induced peroxisomal and mitochondrial fission2,3. Computational protein sequence analysis revealed a high propensity for HD1 to adopt an amphipathic helix, where polar and hydrophobic residues disproportionate to two distinct halves of the structure (Fig. 2A). Amphipathic helices with a similar distribution of polar and apolar amino acids are known to interact with membranes and induce membrane bending, curvature, and remodelling24,25. Recombinant full-length GDAP1 turned out to be not soluble when expressed in either bacteria11 or even in insect cells (data not shown). However, we have previously demonstrated that in vitro translated full-length GDAP1 integrates readily into liposomes2. We now made use of this experimental approach to test the amphipathic characteristics of GDAP1’s HD1. We incubated in vitro translated GDAP1 with liposomes of different compositions26 and analyzed the liposome morphology by negative staining (Fig. 2B). Liposomes without any addition of in vitro translated protein have a round shape, which was quantified by morphometric analysis (Fig. 2C). The addition of GDAP1 T288X lacking both HD1 and TMD served as negative control as this protein is not targeted to membranes2,3. In contrast to the control, in vitro translated full-length GDAP1 added to liposomes mimicking outer mitochondrial or peroxisomal membranes induced membrane outfoldings and significantly deformed the liposomes, while pure phosphatidylcholine liposomes are not deformed by the addition of the same translation mix (Fig. 2B,C). To investigate the role of HD1’s amphipathic pattern for this membrane remodelling activity of GDAP1, we also translated in vitro full-length GDAP1 with a scrambled HD1 (HD1scr) to break the amphipathic structure (Fig. 2A). Similar to the negative-control, HD1scr did not deform the liposome surface of all three membrane-compositions (Fig. 2B,C).

GDAP1 tubulates liposomes dependent on the HD1 and the lipid composition.
(A) Helical wheel representation of residues of the HD1 reveals the amphipathic pattern of hydrophilic and hydrophobic amino acids. This pattern is broken up by scrambling the primary sequence of the HD1 (HD1scr). (B) Electron microscopy of negatively stained liposomes of phosphatidylcholine (PC) or of lipid compositions resembling the mitochondrial outer membrane (Mito) or peroxisomal membrane (Peroxi) were incubated with in vitro translated GDAP1, GDAP1 HD1scr, GDAP1 lacking HD1 and TMD (GDAP1 T288X), or were left untreated. All liposomes have primarily multilamellar appearances. Only the addition of GDAP1 to Mito- and Peroxi-liposomes caused tubulation (arrows). Scale bars: 50 nm. (C) Per preparation 16 to 25 electron micrographs were taken blindly. On electronic pictures all discrete liposomes were selected automatically and the circularity of the objects was determined. The graph depicts the mean and the s.e.m. of all liposomes per condition (n = 79 to 197) from independent preparations, paired t-test ***P-value < 0.0005.
Discussion
We provide here first experimental evidence that GDAP1 has a GSH-conjugation activity typical for theta-class GSTs. While full-length GDAP1 cannot be purified in soluble form from E. coli or insect cell (not shown), C-terminal truncated recombinant GDAP1 proteins without the C-terminal transmembrane domain (TMD) are readily expressed and soluble. More interestingly, we found that only GDAP1 lacking both its C-terminal TMD and HD1 has GSH-conjugating activity. By size-exclusion chromatography (SEC) and immunoprecipitation experiments we confirm that GDAP1 with HD1 but without the TMD also forms dimers. We conclude that loss of GST-activity in the presence of HD1 is not caused by a protein dimerization deficit and hypothesize that HD1 might have an auto-inhibitory function in regulating the enzyme activity of GDAP1.
To understand the potential role of HD1 in GDAP1 we performed in silico analysis, which predicted an amphipathic pattern for HD1, a structure known for its membrane-remodeling activities. Indeed, the addition of in vitro translated full-length GDAP1 to liposomes mimicking the peroxisomal or outer mitochondrial membrane composition lead to a deformation of these liposomes. Incubation oft the same liposomes with GDAP1 with a scrambled HD1 sequence or GDAP1 lacking the TMD does not alter the liposomal shape. This corroborates our previous findings showing that the HD1 is essential to mediate GDAP1-induced mitochondrial and peroxisomal fission3,4. However, we could not link the membrane remodeling capacity of GDAP1 with GDAP1’s GST activity as the in vitro translated full-length GDAP1 did not prove to have GST activity (not shown) and the GST-active recombinant GDAP1 is not targeted to membranes as it lacks the tail-anchor domain2.
Implementing our new findings together with results from previous reports2,3,4,10,11 into a hypothetical working model, we suggest that GDAP1 function may be mechanistically regulated by adopting two different conformations, an active and inactive state (Fig. 3). In the active state, GST activity is not inhibited, owing to the fact that HD1 is exposed and dips into the cytosolic leaflet of the MOM or peroxisomal membrane to mediate membrane curvature. In contrast, in the inactive state the interaction with the membrane is lost and HD1 acts as an autoinhibitory domain locking the N-terminal cytosolic domains of GDAP1 in a fission- and GST-inactive conformation. We propose that GDAP1 switches between these distinct molecular conformations.

Two-conformation molecular switch model of GDAP1: Active and inactive states.
The fission factor GDAP1 is anchored with its C-terminal TMD into the mitochondrial outer membrane or peroxisomal membrane. Its N-terminal GST-domains and the HD1 are cytosolic. We hypothesize that GDAP1’s fission activity is regulated by its enzymatically active GST-portion and dependent on HD1. In the inactive state GDAP1 is not capable of inducing fission. Instead, HD1 is blocking the GST activity in an autoinhibitory fashion (left). In the active state, GDAP1 induces fission (right) dependent on the amphipathic pattern of HD1, which induces membrane curvature. Wild type GDAP1 acts as a molecular switch between the active and the inactive state, depending on specific stimuli. dmGDAP1 preferentially adopt the active conformation resulting in toxic hyper-fission activity, whereas rmGDAP1 have lost or markedly reduced fission activity as they preferentially adopt the inactive state. Experimental evidence for the different states is currently lacking due to the absence of high-resolution structural data.
In the absence of high-resolution atomic structures of GDAP1, experimental prove for our proposed model is currently lacking. Notwithstanding, evidence exists that is consistent with our model, including the observed properties of point mutated disease-associated forms of GDAP1. Recessively inherited mutations cluster within the coding region of the N-terminal, cytosolic GST-N, GST-C, and interdomain of GDAP16, suggesting that the function of GDAP1’s GST domain is impaired in recessively inherited mutant forms. We speculate that GDAP1 expression may be protective by its GST activity, and GSTs were proposed to exert such functions18,19,20. However, our attempts to determine GST activity of disease-associated mutants failed, as all tested recombinant mutated GDAP1 proteins isolated from insect cells were not soluble, indicating non-functional folding of the mutants.
Expressing rmGDAP1 is associated with a loss or markedly reduced fission capacity, while dmGDAP1 still induce fission but block or delay mitochondrial fusion1,5. Based on our model we speculate that mutant forms of GDAP1 associated with CMT are likely to favour one of the two protein conformations. dmGDAP1 protein might be in a pro-active, fission-inducing conformation, resulting in a fragmented mitochondrial network, whereas the rmGDAP1 remain in the inactive, fission- and GST-inhibited conformation4,9. This suggests that GDAP1 function relies on an activating stimulus to switch between conformations. We have previously described a similar activation switch for the cytosolic GDAP1-paralogue GDAP1like1, which translocates to mitochondria and integrates into the MOM upon changes of the cellular redox conditions, inducing mitochondrial fission17. In analogy we favour the interpretation that GDAP1’s GST domains function as a redox sensor, rather than a bona fide GST enzyme, changing its conformational state upon activating stimuli, which regulates the release of the autoinhibitory HD1 domain to execute fission by interaction with the organelle’s outer leaflet.
Besides the induction of mitochondrial and peroxisomal fission, GDAP1 has been linked to the regulation of intracellular Ca(2+) homeostasis27,28. Interestingly, loss of GDAP1 expression can be modified by junctophilin-1, which restores the store-operated Ca(2+) entry (SOCE)27. Although the morphological changes of mitochondria are not sufficient to explain changes in intracellular Ca(2+) homeostasis29, intracellular Ca(2+) levels and redox conditions are very tightly linked30. GDAP1 might function as a mitochondrial redox sensor and transducer of ROS signals to SOCE. Similarly, mitochondrial fusion factor mitofusin-2 (MFN2), which is mutated in axonal forms of CMT (OMIM: 609260), influences the entry of Ca(2+) when mitochondria are depolarized31. Furthermore, oxidized glutathione levels directly influence MFN2 fusion activity32. Taken together these results point to a common theme in CMT caused by mutations in GDAP1 or MFN2, which converge on sensing of redox stress conditions and result in the regulation of mitochondrial morphology and intracellular Ca(2+) levels. However the precise conditions stimulating these proteins in vivo and the resulting molecular and cellular reactions remain to be elucidated.
Materials and Methods
Cell culture
HEK-293T cells were cultured and transfected with Lipofectamine 2000 (Invitrogen) as described previously1,3. The insect cells Sf21 were cultured, maintained and infected as described23.
Plasmids and antibodies
Human GDAP1 (hGDAP1) missense mutations and truncations were cloned into the pcDNA3.1 expression vector as described previously1,2. The constructs GDAP1 T288X and GDAP1 T3188X and their mutations were cloned into vector pFL from the MultiBac system. All constructs comprise a hexa-histidine tag at their C-terminus23. For Western blotting and immunocytochemistry, anti-mouse GDAP1 (Pineda, Berlin, Germany) and anti-human GDAP1 (Pineda, Berlin, Germany) were used as described1,9.
Recombinant protein production
Proteins were expressed as described in ref. 23. The constructs were all purified in the same 2-step manner. Cells were solubilized in buffer A (25 mM Tris pH8, 200 mM NaCl) and lyzed by freeze-thaw. Soluble and insoluble fractions were separated by ultra-centrifugation (1 h at 45 kRPM in Beckman Ti70 rotor). The supernatant was then loaded to a pre-equilibrated Talon affinity column (Clontech, Stain-Germain-en-Laye, France) and eluted with a gradient changing to buffer B (25 mM Tris pH8, 200 mM NaCl, 250 mM imidazole). The relevant fractions were pooled and supplemented with 2 mM DTT. Finally, the concentrated fractions were purified with a pre-equilibrated Superdex 200 10/300 (GE Healthcare, Uppsala, Sweden) in buffer C (20 mM Tris pH 8.0, 100 mM NaCl, 1 mM DTT, 1 mM EDTA). The in vitro translation of GDAP1 wildtype and mutant forms was performed using the TNT T7 Quick Coupled reticulocyte system (Promega, Madison, USA) as described previously2.
Immunoprecipitation
30 μl wet Protein A-Sepharose beads (GE Healthcare, Uppsala, Sweden) were incubated overnight with 5 μl GDAP1 antibody serum. Transfected HEK-293T were harvested in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1:100 Protease inhibitor cocktail (Sigma, St. Louis, USA)), centrifuged for 5 min at 800 g and the cleared supernatant was than incubated with the antibody coupled beads for 2 h. Beads were washed four times with lysis buffer, and subsequently the samples were boiled at 95 °C in SDS-loading buffer and subjected to immunoblotting analysis.
GST activity measurements
For all GST activity assays 50 μg of recombinant protein (in buffer 20 mM Tris pH 8.0, 100 mM NaCl, 1 mM DTT, 1 mM EDTA) in a total reaction volume of 200 μl using the conditions listed in Supplementary Table 1 (see Supplementary Table 1) were used33,34. Crude rat liver extract served as positive control and buffer only as negative control for any assay. The absorbance was measured every 30 sec in a multi-well plate Photometer at room temperature for ten to twenty minutes (SpectraMax190, Molecular Devices, Sweden).
Liposome generation and analysis
Liposomes were prepared using 5 mM stock solutions in methanol:chloroform (1:4) of phosphtidycholine (PC, Sigma, St. Louis, USA), L-α-phosphatidylethanolamine (PE), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (18:1; PS), L-α-phosphatidylinositol (PI), cardiolipin (CA). Lipids were purchased from Avanti Polar Lipids Inc., if not indicated differently. To prepare liposomes in the desired composition, 20 μl lipid-mixture (PC = 20 μl PC; Peroxi = 11 μl PC, 6 μl PE, 1 μl PS, 1 μl PI, 1 μl CL26; Mito = 10.8 μl PC, 4 μl PE, 5.2 μl CL35 were dried under vacuum for 2 h. Lipids were resuspended in 400 μl buffer (10 mM HEPES pH 7.4, 1 mM EDTA, 250 mM sucrose) and passed ten times through two 100-nm pore size polycarbonate filters (Avestin). 50 μl liposome solution were incubated with 10 μl in vitro translate or were left untreated for 4 hours on ice, mixed 1:1 with 2% phosphotungstic acid (pH 7.2) and immediately placed on Formvar-coated nickel grids (200 mesh, PLANO) for 4 minutes. The liquid was removed and the grids were dried for 2 hours under vacuum and examined in blinded fashion with a Zeiss EM 109S electron microscope (Zeiss, Oberkochen, Germany). On the electronic pictures all separate liposomes were selected with the magic wand tool in Adobe Photoshop and the circularity of the objects was determined.
Software
Data analysis was performed using Excel (Microsoft) and the amphipathic pattern was determined by the modeling software http://cti.itc.virginia.edu/~cmg/wheel/wheelApp.html.
Additional Information
How to cite this article: Huber, N. et al. Glutathione-conjugating and membrane-remodeling activity of GDAP1 relies on amphipathic C-terminal domain. Sci. Rep. 6, 36930; doi: 10.1038/srep36930 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Niemann, A., Ruegg, M., La Padula, V., Schenone, A. & Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol. 170, 1067–1078, doi: 10.1083/jcb.200507087 (2005).
Wagner, K. M., Ruegg, M., Niemann, A. & Suter, U. Targeting and function of the mitochondrial fission factor GDAP1 are dependent on its tail-anchor. PloS one 4, e5160 (2009).
Huber, N., Guimaraes, S., Schrader, M., Suter, U. & Niemann, A. Charcot-Marie-Tooth disease-associated mutants of GDAP1 dissociate its roles in peroxisomal and mitochondrial fission. EMBO Rep. 14, 545–552, doi: 10.1038/embor.2013.56embor201356 [pii] (2013).
Niemann, A., Wagner, K. M., Ruegg, M. & Suter, U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiology of disease 36, 509–520 (2009).
Pedrola, L. et al. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Human molecular genetics 14, 1087–1094, doi: ddi121 [pii]10.1093/hmg/ddi121 (2005).
Cassereau, J. et al. A locus-specific database for mutations in GDAP1 allows analysis of genotype-phenotype correlations in Charcot-Marie-Tooth diseases type 4A and 2K. Orphanet J Rare Dis 6, 87, doi: 1750-1172-6-87 [pii]10.1186/1750-1172-6-87 (2011).
Suter, U. & Scherer, S. S. Disease mechanisms in inherited neuropathies. Nature Rev. Neurosci. 4, 714–729 (2003).
Niemann, A., Berger, P. & Suter, U. Pathomechanisms of mutant proteins in Charcot-Marie-Tooth disease. Neuromolecular Med. 8, 217–242 (2006).
Zimon, M. et al. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 77, 540–548, doi: WNL.0b013e318228fc70 [pii]10.1212/WNL.0b013e318228fc70 (2011).
Noack, R. et al. Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Human molecular genetics 21, 150–162, doi: ddr450 [pii]10.1093/hmg/ddr450 (2012).
Shield, A. J., Murray, T. P. & Board, P. G. Functional characterisation of ganglioside-induced differentiation-associated protein 1 as a glutathione transferase. Biochem Biophys Res Commun 347, 859–866, doi: S0006-291X(06)01421-5 [pii]10.1016/j.bbrc.2006.06.189 (2006).
Cuesta, A. et al. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet 30, 22–25 (2002).
Marco, A., Cuesta, A., Pedrola, L., Palau, F. & Marin, I. Evolutionary and Structural Analyses of GDAP1, Involved in Charcot-Marie-Tooth Disease, Characterize a Novel Class of Glutathione Transferase-Related Genes. Mol Biol Evol. 21, 176–187 (2004).
Hayes, J. D., Flanagan, J. U. & Jowsey, I. R. Glutathione transferases. Annu Rev Pharmacol Toxicol. 45, 51–88, doi: 10.1146/annurev.pharmtox.45.120403.095857 (2005).
Oakley, A. Glutathione transferases: a structural perspective. Drug Metab Rev. 43, 138–151, doi: 10.3109/03602532.2011.558093 (2011).
Sheehan, D., Meade, G., Foley, V. M. & Dowd, C. A. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. The Biochemical journal 360, 1–16 (2001).
Niemann, A. et al. The Gdap1 knockout mouse mechanistically links redox control to Charcot-Marie-Tooth disease. Brain: a journal of neurology 137, 668–682, doi: 10.1093/brain/awt371 (2014).
Raza, H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. The FEBS journal 278, 4243–4251, doi: 10.1111/j.1742-4658.2011.08358.x (2011).
Goto, S. et al. Glutathione S-transferase pi localizes in mitochondria and protects against oxidative stress. Free radical biology & medicine 46, 1392–1403, doi: 10.1016/j.freeradbiomed.2009.02.025 (2009).
Raza, H., Robin, M. A., Fang, J. K. & Avadhani, N. G. Multiple isoforms of mitochondrial glutathione S-transferases and their differential induction under oxidative stress. The Biochemical journal 366, 45–55, doi: 10.1042/BJ20020533 (2002).
Robin, M. A., Prabu, S. K., Raza, H., Anandatheerthavarada, H. K. & Avadhani, N. G. Phosphorylation enhances mitochondrial targeting of GSTA4-4 through increased affinity for binding to cytoplasmic Hsp70. The Journal of biological chemistry 278, 18960–18970, doi: 10.1074/jbc.M301807200 (2003).
Pla-Martin, D. et al. Silencing of the Charcot-Marie-Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiology of disease 55, 140–151, doi: 10.1016/j.nbd.2013.03.010 (2013).
Bieniossek, C., Richmond, T. J. & Berger, I. MultiBac: multigene baculovirus-based eukaryotic protein complex production. Curr Protoc Protein Sci Chapter 5, Unit 5 20, doi: 10.1002/0471140864.ps0520s51 (2008).
Antonny, B. Mechanisms of membrane curvature sensing. Annual review of biochemistry 80, 101–123, doi: 10.1146/annurev-biochem-052809-155121 (2011).
Shen, H., Pirruccello, M. & De Camilli, P. SnapShot: membrane curvature sensors and generators. Cell 150, 1300, 1300 e1301–1302, doi: 10.1016/j.cell.2012.08.017 (2012).
Opalinski, L., Kiel, J. A., Williams, C., Veenhuis, M. & van der Klei, I. J. Membrane curvature during peroxisome fission requires Pex11. Embo J 30, 5–16, doi: 10.1038/emboj.2010.299 emboj2010299 [pii] (2011).
Pla-Martin, D. et al. Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Human molecular genetics 24, 213–229, doi: 10.1093/hmg/ddu440 (2015).
Barneo-Munoz, M. et al. Lack of GDAP1 induces neuronal calcium and mitochondrial defects in a knockout mouse model of charcot-marie-tooth neuropathy. PLoS Genet 11, e1005115, doi: doi: 10.1371/journal.pgen.1005115 (2015).
Frieden, M. et al. Ca(2+) homeostasis during mitochondrial fragmentation and perinuclear clustering induced by hFis1. The Journal of biological chemistry 279, 22704–22714, doi: 10.1074/jbc.M312366200 (2004).
Nunes, P. & Demaurex, N. Redox regulation of store-operated Ca2+ entry. Antioxid Redox Signal 21, 915–932, doi: 10.1089/ars.2013.5615 (2014).
Singaravelu, K. et al. Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. The Journal of biological chemistry 286, 12189–12201, doi: 10.1074/jbc.M110.174029 (2011).
Shutt, T., Geoffrion, M., Milne, R. & McBride, H. M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 13, 909 -915, doi: 10.1038/embor.2012.128 embor2012128 [pii] (2012).
Habig, W. H., Pabst, M. J. & Jakoby, W. B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. The Journal of biological chemistry 249, 7130–7139 (1974).
Widersten, M. & Mannervik, B. Glutathione transferases with novel active sites isolated by phage display from a library of random mutants. Journal of molecular biology 250, 115–122, doi: 10.1006/jmbi.1995.0362 (1995).
Montessuit, S. et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142, 889–901, doi: 10.1016/j.cell.2010.08.017 (2010).
Acknowledgements
We thank all members of the Suter and Berger laboratories for helpful discussions. We thank Helge Ewers for technical support. This work was supported by the Swiss National Science Foundation [US] and by the European Commission Framework Programme 7 ComplexINC project [contract nr. 279039] [IB].
Author information
Authors and Affiliations
Contributions
All authors were involved in experimental design. N.H., C.B., K.M.W., H.P.E. and A.N. performed the experiments. All authors were involved in data interpretation and analysis. N.H., U.S., I.B. and A.N. wrote the manuscript with input from all authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Huber, N., Bieniossek, C., Wagner, K. et al. Glutathione-conjugating and membrane-remodeling activity of GDAP1 relies on amphipathic C-terminal domain. Sci Rep 6, 36930 (2016). https://doi.org/10.1038/srep36930
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36930
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
