
Abstract
Two new grayanoids, rhodomollin A (1) and rhodomollin B (2), possessing an unprecedented D-homo grayanane carbon skeleton, were isolated from the fruits of Rhododendron molle. The structures of 1 and 2 were fully characterized using a combination of spectroscopic analyses and X-ray crystallography. Rhodomollin B (2) exhibited modest activity against influenza virus A/95-359, with an IC50 value of 19.24 μM.
Similar content being viewed by others

Introduction
Grayanoids are characteristic metabolites of Rhododendron plants and possess diverse structures and significant biological activities1. In particular, grayanoids with a wide spectrum of biological activities, such as sodium-channel-modulating2, analgesic3, sedative4, and insect antifeedant activities5, have been isolated from plants of the Rhododendron genus in recent years. Ten types of grayanane-related carbon skeletons have been reported, including grayanane (A-nor-B-homo ent-kaurane)6, 1,5-secograyanane7, 3,4-secograyanane8, 9,10-secograyanane9, 1,10:2,3-disecograyanane10, leucothane (A-homo-B-nor grayanane)11, kalmane (B-homo-C-nor grayanane)12, 1,5-seco kalmane13, micranthane (C-homo grayanane)14, and mollane (C-nor-D-homo grayanane)15. We previously reported mollolide A, a grayanoid with a new 1,10:2,3-disecograyanane skeleton10, and mollanol A, the first example with a C-nor-D-homo grayanane carbon skeleton15, from Rhododendron molle G. Don, a plant that is used in traditional Chinese medicine as an anodyne and anesthetic16. In our continuing efforts to identify structurally interesting and biologically important grayanoids, two new grayanoids, rhodomollin A (1) and rhodomollin B (2), possessing an unprecedented D-homo grayanane (featured a 5/7/6/6-fused ring system) carbon skeleton, were isolated from the fruits of R. molle. Herein, the isolation, structural elucidation, and evaluation of the anti-viral activity of these compounds are discussed.
Results and Discussion
Rhodomollin A (1) was obtained as a colorless crystal, and its molecular formula was determined to be C20H30O5 with six degrees of unsaturation based on HRESIMS analysis (m/z 373.1988 [M + Na]+, calcd 373.1985), consistent with the 1H and 13C NMR data. The IR spectrum included a strong absorption band at 3424 cm−1, indicative of the presence of hydroxy group(s). Analysis of the 13C NMR (DEPT) data led to the assignment of four methyls, three methylenes, eight methines (three oxygenated, two olefinic carbons), and five quaternary carbons (three oxygenated) (see Table 1). The two olefinic carbons accounted for only one of the six degrees of unsaturation; thus, five rings should be present. Detailed analysis of the 1H and 13C data of 1 indicated that 1 shares partial structural features (rings A, B and C in Fig. 1) with previously reported grayanane-type diterpenoids.

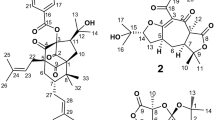
Structures of compounds 1 and 2.
The two gem-dimethyl singlets (δH 1.51, H3-18; 1.47, H3-19) were chosen as the starting point for the structural elucidation of ring A. The HMBC correlations from H3-18/H3-19 to C-3/C-4/C-5, from H-2 to C-5, and from H-3 to C-1 (see Fig. 2) and the assignments of fragment a [C(1)H−C(2)H−C(3)H] by 1H−1H COSY and HSQC established the five-membered ring A.

Selected 1H-1H COSY, HMBC, and NOESY correlations for compound 1.
Ring B was elucidated by the HMBC correlations from H-6 to C-1, C-5 and C-10, from H3-20 to C-1 and C-10, and from H-2 to C-1, C-5, and C-10. Because the five oxygen atoms in the molecular formula accounted for the six oxygenated carbons in the 13C-NMR spectrum and H-6 was correlated to C-10 in the HMBC spectrum, ring B was determined to be a furan ring formed through a C6-O-C10 oxygen bridge and fused with ring A at C-1 and C-5. The HMBC correlations from H3-20 to C-9 and from H2-7 to C-8/C-9 together with spin system b, C(6)H-C(7)H2, indicated that ring C shares the C6-O-C10 oxygen bridge with ring B to form a pyran ring.
The HMBC correlations from H-14 to C-8 and C-9 and from H-9 to C-14, together with spin system c elucidated from the 1H-1H COSY and HSQC spectra, established a six-membered ring (ring D in Fig. 1). In addition, the HMBC correlations from H2-15 to C-8, C-9, and C-14 indicated the linkage of C-15 and C-8. The HMBC correlations from H-12 to C-16 and C-15 confirmed the connectivity of C-16 and C-12. Thus, another six-membered ring (ring E) was established. Subsequently, the HMBC correlations from H3-17 to C-12, C-15, and C-16 unambiguously placed CH3-17 on C-16.
Consequently, the planar structure of 1 was determined and features a 5/5/6/6/6-fused ring system. Rings D and E compose a bicyclo[2.2.2]octane ring system in which C-8 and C-12 are bridged by C-15 and C-16. This is the first example of a D-homo grayanane carbon skeleton.
The relative configurations were determined by NOESY correlations (Fig. 2). NOESY correlations of H-3/H-1 and OH-3/H3-19 revealed the α-orientation of H-1 and H-3. NOESY correlations between H-1 and H-14 confirmed that the C14-C8 bond was also α-oriented, indicating that bond C8-C15 is β-configured. Additionally, H-12 must adopt an α-orientation in the rigid bicyclo[2.2.2]octane ring system formed by rings D and E. NOESY correlations of H-7b/H-9, H-9/H-15a, and H-9/OH-16 indicated that H-9, OH-16, H-15a, and H-7b are on the same side with a β-configuration.
The NOESY correlations of OH-5/H-1 and OH-5/H3-18 suggested that the two five-membered rings A and B were cis-fused to form a bicyclo[3.3.0]octane. The cis-fusion of rings A and B is preferred in bicyclo[3.3.0]octane because the trans-fusion of two five-membered rings would involve considerable torsional strain, with a very large enthalpy difference (cis to trans 29.6 kJ/mol)17. Due to a lack of NOE evidence, the relative configuration of the C6-O-C10 oxygen bridge could not be assigned directly by NOESY. Fortunately, a high-quality single crystal of 1 was obtained from a methanol-water solvent system. The X-ray crystallographic (Cu Kα radiation) data (Table S1–S4) corroborated the planar structure and the relative configuration of 1 and further allowed the assignment of its absolute configuration as 1R, 2R, 3R, 5S, 6R, 8R, 9R, 10S, 12R, 16S [with a Flack parameter of 0.05 (10)] (see Fig. 3). The crystallographic data for 1 have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number 1445432.

X-ray structure of compound 1.
Rhodomollin B (2) was obtained as white amorphous powder. Its molecular formula, C20H30O5, as deduced from HRESIMS, is identical to that of 1. The UV, IR, and NMR spectral data of 2 also resembled those of 1, except that compared with 1, the 13C NMR signals for C-15 and C-17 in 2 were downshifted 1.5 ppm and upshifted 2.3 ppm, respectively. Careful analysis of the spectroscopic data allowed us to conclude that its planar structure was identical to that of 1.
The NOESY correlations of H-1/H-3, H-1/H-14, H-9/H-7b, H-9/H-15a, and OH-5/H3-18 revealed that the two compounds share the same relative configurations in these positions, except the stereochemistry at C-16. The relative configuration of OH-16 was then confirmed to be α-oriented on the basis of the NOESY correlation between H-9 (β) and H3-17, indicating that 2 is a C-16 diastereomer of 1. Thus, the structure of 2 was established and named rhodomollin B.
Rhodomollins A (1) and B (2) represent a new tetracyclic diterpene carbon skeleton with an unprecedented D-homo grayanane (featured a 5/7/6/6-fused ring system) carbon skeleton. We have named this new skeleton “rhodomollane”. Compound 1 possesses an unusual β-16-OH. The stereochemistry at C-16 in grayanoids is highly conserved. A likely biosynthetic pathway is proposed in Fig. 4. Compounds 1 and 2 share a common precursor i, which undergoes a C13-alkyl shift to form a carbocation center at C-1618. The carbocation center at C-16 can then be attacked by H2O from two sites to form diastereomers 1 and 2.

Proposed biosynthetic pathway for 1 and 2.
The anti-viral activities of rhodomollins A (1) and B (2) were assessed in vitro in A/95-359 influenza virus-infected MDCK cells. Rhodomollin B (2) exhibited modest activity against influenza virus A/95-359, with an IC50 value of 19.24 μM, whereas rhodomollin A (1) was inactive (IC50 > 100 μM).
Materials and Methods
General experimental procedures
Optical rotations were measured with a PE model 343 polarimeter. CD spectra were recorded on a JASCO-815 CD spectrometer. A Nicolet 5700 FT-IR microscope instrument was used to record IR spectra. NMR spectra were obtained on INOVA-500 spectrometer. ESIMS were measured with an Agilent 1100 Series LC/MSD Trap mass spectrometer. HRESIMS data were measured using an Agilent 6520 Accurate-Mass Q-TOF LC/MS spectrometer. X-ray experiments were carried on a Gemini E X-ray single crystal diffractometer. A preparative Shimadazu LC-6AD HPLC equipped with SPD-20A and RID-10A detectors (Kyoto, Japan) along with an YMC ODS-A column (250 × 20 mm, 5 μm, Kyoto, Japan) were used to purify the compounds. Macroporous resin (D101 type, The Chemical Plant of NanKai University, China), MCI gel, Mitsubishi chemical corporation, Sepherdex LH-20, GE chemical corporation, Si gel (160–200, 200–300 mesh, Qingdao Marine Chemical Factory, China) and ODS (50 μm, Merck, Germany) were used for column chromatography (CC). TLC was carried out with precoated Si gel plates (Qingdao Marine Chemical Factory, China). Spots were visualized by spraying with 10% EtOH-sulfuric reagent; see supplementary information S4.
Plant material
The fruits of Rhododendron molle were collected in Guangxi Province of China, in 2012. It was authenticated by Guang-Zhao Li, a professor of Guangxi Institute of Botany. A voucher specimen (ID-s-2445) was deposited in the herbarium of our institute (Institute of Materia Medica, Chinese Academy of Medical Sciences); see supplementary information S4.
Extraction and isolation
Extracts from the dried fruits of Rhododendron molle (100 kg) were obtained (2 h per extraction) with EtOH-H2O (95:5, v/v) under conditions of reflux. The extract was suspended in 30 L of H2O, and then partitioned with petroleum ether, CH2Cl2, EtOAc and MeOH (three times with 15 L each). The EtOAc fraction was then further separated on a macroporous resin column and eluted in a gradient of H2O:EtOH (70:30, 40:60, 5:95, v/v) in order of increasing concentrations of EtOH. The 30% EtOH fraction was further resolved on a MCI gel column and eluted in a gradient of MeOH:H2O (1:9–10:0, v/v) to obtain 15 fractions (EM1–EM15). Fraction EM9 was purified by Sephadex LH-20 column to obtain a terpenoid-containing fraction EM9G1 (9 g), which was further loaded onto an Si gel column and eluted in a gradient of CH2Cl2:MeOH (20:1–1:2, v/v) to obtain 10 fractions (EM9G1L1–EM9G1L10). EM9G1L6 (0.68 g) was purified by preparative HPLC and semi-preparative HPLC to yield 1 (6.0 mg). EM9G1L7 (0.55 g) was purified by preparative HPLC and semi-preparative HPLC to yield 2 (2.2 mg); see supplementary information S5.
Rhodomollin A (1)
Colorless crystals; [α]20D + 7.1 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 204.8 (2.86) nm; IR(KBr) νmax 3424, 2960, 2932, 1645, 1463, 1418, 1381, 1139, 1057, 1029, 984, 907, 875, 817, 798, 730, 665, 635 cm−1. For 1H and 13C NMR spectroscopic data, see Table 1; HRESI-MS 373.1986 (calcd for C20H30NaO5, 373.1985); see supplementary information S6–S19.
Rhodomollin B (2)
White powder; [α]20D + 8.5 (c 0.14, MeOH); UV (MeOH) λmax (log ε) 204.0 (2.9) nm; IR(KBr) νmax 3374, 2931, 2872, 1597, 1443, 1379, 1123, 1081, 1050, 1129, 926, 910, 816, 732, 634 cm−1. For 1H and 13C NMR spectroscopic data, see Table 1; HRESI-MS 373.1986 (calcd for C20H30NaO5, 373.1985); see supplementary information S20–S33.
Anti-influenza A Assays
Madin-Darby Canine Kidney (MDCK) cells and influenza A (A/Hanfang/359/95, H3N2) were from the Institute of Virology, Chinese Academy of Preventive Medicine. Confluent Madin-Darby Canine Kidney (MDCK) cells were infected with influenza A (A/Hanfang/359/95, H3N2) in 96-well microplates. After 2 h of viral adsorption (37 °C), the monolayers were washed by PBS and incubated with or without test compounds in the maintenance medium (37 °C). Cytopathogenic effect (CPE) caused by viral invasion was evaluated when the viral control group reached a level of 4 and the antiviral activity of test compounds was determined according to the Reed and Muench method19,20,21.
The cytotoxicity of compounds in the presence of MDCK cells were monitored by CPE. MDCK cells (2.5 × 104/well) were plated into a 96-well plate. A total of 24 h later, the monolayer cells were incubated in the presence of various concentrations of test compounds. The cells were cultured for 48 h at 37 °C and 5% CO2 in a carbon-dioxide incubator. CPE were counted on the infected MDCK cells. Median toxic concentration (TC50) values of test compounds were calculated by the method of Reed and Muench. Compounds 1 and 2 showed no cytotoxicicy in this assay (TC50 > 100 μM).
Additional Information
How to cite this article: Li, Y. et al. Rhodomollins A and B, two Diterpenoids with an Unprecedented Backbone from the Fruits of Rhododendron molle. Sci. Rep. 6, 36752; doi: 10.1038/srep36752 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Li, Y., Liu, Y. B. & Yu, S. S. Grayanoids from the Ericaceae family: structures, biological activities and mechanism of action. Phytochem. Rev. 12, 305–325 (2013).
Maejima, H. et al. Grayanotoxin binding and unbinding to D4S6 of Nav1.4 sodium channel as revealed by improved estimation of toxin sensitivity. J. Biol. Chem. 278, 9464–9471 (2003).
Li, Y. et al. Antinociceptive grayanoids from the roots of Rhododendron molle. J. Nat. Prod. 78, 2887–2895 (2015).
Wang, S. et al. Highly acylated diterpenoids with a new 3,4-secograyanane skeleton from the flower buds of Rhododendron molle. Org. Lett. 12, 1560–1563 (2010).
Klocke, J. A. et al. Grayanoid diterpene insect antifeedants and insecticides from Rhododendron molle. Phytochemistry 30, 1797–1800 (1991).
Narayanan, P. et al. Crystal and molecular structure of grayanotoxin-I. Tetrahedron Lett. 11, 3943–3944 (1970).
Fushiya, S., Hikino, H. & Takemoto, T. Stereostructure of grayanol A and B, diterpenoids of Leuc−othoe grayana. Tetrahedron Lett. 15, 183–186 (1974).
Li, C. H. et al. Novel polyesterified 3,4-seco-grayanane diterpenoids as antifeedants from Pieris formosa. Org. Lett. 12, 2426–2429 (2010).
Wu, Z. Y. et al. Lyonin A, a new 9, 10-Secograyanotoxin from Lyonia ovalifolia. Chem. Biodivers. 8, 1182–1187 (2011).
Li, Y. et al. Mollolide A, a diterpenoid with a new 1,10:2,3-disecograyanane skeleton from the roots of Rhododendron molle. Org. Lett. 15, 3074–3077 (2013).
Furusaki, A., Hamanaka, N. & Miyakoshi, H. Leucothol A, an anthraditerpenoid from Leucothoe grayana Max. Chem. Lett. 1, 783–786 (1972).
Burke, J. W. et al. Kalmanol, a pharmacologically active diterpenoid with a new ring skeleton from Kalmia angustifolia L. J. Am. Chem. Soc. 111, 5831–5833 (1989).
Zhou, S. Z. et al. Diterpenoids from the flowers of Rhododendron molle. J. Nat. Prod. 77, 1185–1192 (2014).
Zhang, M. K. et al. Micranthanone A, a new diterpene with an unprecedented carbon skeleton from Rhododendron micranthum. Org. Lett. 15, 3094–3097 (2013).
Li, Y. et al. Mollanol A, a diterpenoid with a new C-nor-D-homograyanane skeleton from the fruits of Rhododendron molle. Org. Lett. 16, 4320–4323 (2014).
Chen, J. S. & Zhen, S. Chinese Poisonous Plants. Ch. 34, 232 ( Zhang, C. L., 1987).
Kendhale, A. M. et al. A rigid bicyclo[3.3.0]octane (octahydropentalene): a heavily constrained novel aliphatic template for molecular self-assembly. Tetrahedron Lett. 49, 3056–3059 (2008).
Riehl, R. S. et al. New avenues for the synthesis of ent-kaurene diterpenoids. Tetrahedron 71, 6629–6650 (2015).
Li, Y. P. et al. Synthesis and biological evaluation of heat-shock protein 90 inhibitors: geldanamycin derivatives with broad antiviral activities. Antiviral Chem. Chemother. 20, 259–268 (2010).
Ji, X. Y. et al. Synthesis and antiviral activity of N-phenylbenzamide derivatives, a novel class of enterovirus 71 Inhibitors. Molecules 18, 3630–3640 (2013).
Lv, H. N. et al. Sesquiterpenes from the roots of Illicium oligandrum. J. Asian Nat. Prod. Res. 17, 430–438 (2015).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos. 21572274, 21302226 and 21132009). The authors are grateful to the Department of Instrumental Analysis at our institute for the UV, IR, NMR, and MS spectra measurements.
Author information
Authors and Affiliations
Contributions
S.S.Y. and J.Q. designed the study. S.S.Y. supervised the project. Y.L., Y.B.L., H.M.Y. and Y.L.L. performed the isolation, structure analysis, and discovered the compounds. Y.H.L. performed and analyzed the anti-virus bioassay. H.N.L., S.G.M. and J.Q. performed the quantitative analysis and the HPLC-MS analysis. S.S.Y. and Y.L. wrote the paper. All authors contributed to the discussion and interpretation of the results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, Y., Liu, YB., Yan, HM. et al. Rhodomollins A and B, two Diterpenoids with an Unprecedented Backbone from the Fruits of Rhododendron molle. Sci Rep 6, 36752 (2016). https://doi.org/10.1038/srep36752
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36752
This article is cited by
-
Rhodoterpenoids A‒C, Three New Rearranged Triterpenoids from Rhododendron latoucheae by HPLC‒MS‒SPE‒NMR
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
