
Abstract
Chronic hepatic injury is accompanied by a ductular response that is strongly correlated with disease severity and progression of fibrosis. To investigate whether anti-inflammatory drugs can modulate the ductular response, we treated mice suffering from a steatotic or cholestatic injury with anti-TNF-α antibodies (Infliximab) or glucocorticoids (Dexamethasone). We discovered that Dexamethasone and Infliximab can both modulate the adaptive remodeling of the biliary architecture that occurs upon liver injury and limit extracellular matrix deposition. Infliximab treatment, at least in these steatotic and cholestatic mouse models, is the safer approach since it does not increase liver injury, allows inflammation to take place but inhibits efficiently the ductular response and extracellular matrix deposition. Infliximab-based therapy could, thus, still be of importance in multiple chronic liver disorders that display a ductular response such as alcoholic liver disease or sclerosing cholangitis.
Similar content being viewed by others

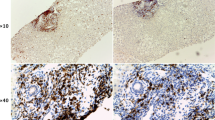

Introduction
During chronic liver disease, progressive destruction of liver parenchyma and/or biliary structures, triggers a complex ductular reaction (or DR) located mainly within the periportal region, which ultimately constitutes the basic regenerative response1. Recent studies show that this DR is intimately associated with fibrosis and cancer2, and that a correlation exists between the extent of liver disease and the magnitude of the accompanying DR3,4.
The DR consists of innumerous bile ductular structures (or reactive-appearing duct-like cells) and intermediate ductal cells, both progenies of the hepatic stem/progenitor cells (HSPCs), with the latter residing in or near the canals of Hering5. Together with these biliary epithelial cell components (BECs), Kupffer cells (KCs), myofibroblasts (MFs), and the extracellular matrix (ECM) act in concert to correctly orchestrate the repopulation and reorganization of the damaged liver tissue1,6.
Tissue repair and regeneration of any organ in response to injury is invariably accompanied by macrophage infiltration7,8. Particularly, macrophages (or inflammatory monocytes) and their cytokine production play a major role in the DR upon chronic liver injury. The number of KCs increases in the liver prior to induction of HSPC proliferation and also spatially co-localize with HSPCs in a niche, suggesting that they may facilitate their recruitment/activation4,9,10 and that inflammation and epithelial repair are intrinsically linked11,12. Macrophages are an important source of soluble factors and cytokines such as tumor necrosis factor- alpha (TNF-α)4. TNF-α modulates a broad range of cellular functions including proliferation, survival, differentiation and apoptosis, and is considered an essential driver of the inflammatory immune response. Data from humans and animal models support a causative role for TNF-α in the pathogenesis of disorders such as alcoholic hepatitis, Crohn’s disease, rheumatoid arthritis, inflammatory bowel disease, and skin diseases, supporting the theoretical rationale for selective TNF inhibition as a beneficial treatment in those patients13,14,15,16. Among the TNF-α antagonists, Infliximab (IFX) is a chimeric monoclonal antibody that binds soluble TNF-α with high affinity and specificity to membrane-bound TNF-α on inflammatory cells and induces inflammatory cell apoptosis13,15,17. Besides anti-TNF-α agents, corticosteroids are widely prescribed anti-inflammatory compounds, which dampen the inflammatory response and are used to treat patients with allergies, asthma, and autoimmune diseases; dexamethasone (DEX) and prednisolone are the most commonly used glucocorticoids. Corticosteroids bind to steroid nuclear receptors, leading to the induction of anti-inflammatory factors and downregulation of pro-inflammatory pathways; in combination, these lead to the suppression of inflammation18.
Therapies that can stop the DR and the accompanying fibrosis are urgently needed. Therefore, we targeted the TNF-α axis by DEX or IFX and evaluated whether this would ameliorate liver injury and subsequent DR and fibrosis in the context of a steatotic (choline-deficient, ethionine-supplement diet: CDE) and a cholestatic (a 3, 5-diethoxycarbonyl-1, 4-dihydrocollidine-containing diet: DDC) disease model.
Results
Evaluation of liver injury upon CDE and DDC treatments
In mice fed with a CDE diet (steatotic), the liver progressively turns to pale brown and has a harder texture than the bloody-red colour of unchallenged livers (Fig. 1A). At day 7, liver injury markers (ALT, AST, and LDH) were elevated while serum total-bilirubin progressively increased until day 14 (Fig. S1). CDE induces steatosis visible as vacuoles by HE staining (Fig. 1B red arrows). An expansion of CK19-positive cells (CK19+ cells) was already evident at day 7 of the CDE regime (Fig. 1B). Although the majority of CK19+-cells at day 7 were located in the periportal region (zone 1), later on, up to 20% CK19+-cells extended into the midlobular region (zone 2). By day 14, the number of CK19+-cells is dramatically increased in CDE-exposed livers, forming arborescence inside the lobule; cells are organized in small clusters, duct-like structures or isolated far from the portal vein area (Fig. 1B).

Histological evaluation of CDE- and DDC-treated livers.
(A) Livers retrieved from unchallenged or mice-fed with a CDE or DDC diet for 14 days. Left lobes are divided for immunohistochemistry studies via embedding-paraffin processing. (B) Liver sections were stained with hematoxylin/eosin and CK19. After 7 days, CDE-treated livers accumulate fat (red arrows), which becomes more pronounced after 2 weeks of treatment, and small oval cells are appearing progressively, whereas porphyrin crystals appearing as brown pigments (red asterix) and stratification of bile ducts were the only striking features observed in livers of DDC-treated mice. In CDE livers, CK19+-cells are dramatically increased in number, forming arborescence inside the lobule. Those cells are organized in small clusters (1), or duct-like structures (2), or isolated (3) far from the PV area. DDC induces morphological alterations of the bile ducts (flattening of cholangiocytes, thickened multi-layered bile duct walls delineating a pseudo lumen) (yellow arrows). All images were taken in 20x original magnification.
In DDC-treated mice (cholestasis), the liver turns to dark-brown (Fig. 1A) and shows a hard liver texture. ALT, AST and LDH at day 7 are increased and plateaued at day 14 (Fig. S1). Given that DDC is a porphyrinogenic drug, biliary obstruction was clearly apparent (Fig. 1B), and serum total-bilirubin drastically increased until day 14 (Fig. S1), resulting in a jaundiced state.
Histological characterization of the ductular reaction induced by CDE and DDC diets
Besides the CK19+ cell expansion upon CDE damage, a DR was illustrated by a clear accumulation of F4/80+ cells, αSMA+ myofibroblasts, and an increase in cross-linked collagen (Sirius red+) and laminin deposition (Fig. S2) indicative of fibrogenesis. A similar outline of the emerging DR was observed with DDC feeding, but this process was more static in terms of the spatial localization of the DR. The CK19+ cells remained primarily in zone 1 with less spreading into the parenchyma compared to CDE treatment (Fig. S3). Accumulation of αSMA+ cells and deposition of laminin were also accompanied by an increase in cross-linked collagen and F4/80+ cells in the DDC-induced DR (Fig. S3). Our results confirm previous studies that distinguish the induction of cholestasis and steatosis in the DDC and CDE mice models, respectively.
Impact of IFX and DEX on CDE-induced ductular reaction
To mimic a potential clinical benefit of IFX and DEX on the DR, we choose to apply these drugs in a therapeutic setting for both models of liver injury (Figs 2A). Administration of IFX or DEX alone has no significant impact on the levels of biochemical parameters, neither on the colour and texture of the livers (Fig. S4), nor on the presence or the number of CK19+ and F4/80+ cells (Fig. S5). IFX or DEX never modulated the appetite of the treated mice; no particular change of fur was noticed nor was special behaviour detected.

Impact of IFX and DEX on CDE diet mediated liver injury parameters and CK19+ cell distribution.
(A) Experimental design: IFX was injected i.p daily from day 7 of CDE treatment whereas DEX was injected from day 9 onwards in a 14 day time course. (B) Quantification of ALT, ALP, and total-bilirubin in sera and hydroxyproline levels in livers from DDC-treated mice +/− DEX or IFX. (C) Representative pictures of immunohistochemistry for CK19 on CDE-treated livers with or without DEX or IFX are shown. The dotted windows in the left panels are magnified and presented in the middle panels. Higher magnifications of CK19+ cells are given in the two right panels. All images were taken in 20x original magnification.All data are mean (+/−SEM) for n = 5/group. *p < 0.05, **p < 0.01, and ***p < 0.001 difference versus CTL. #p < 0.05 and ##p < 0.01 difference versus treated groups (CDE). £p < 0.05 and ££p < 0.01 difference versus co-treated groups (+/−DEX or +/−IFX).
Upon CDE treatment, the liver colour and texture were not modified by DEX or IFX (Fig. S6), nor was the blood volume harvested from those animals. When compared with vehicle-administration (CDE-treated), DEX administration significantly dimmed the high ALT levels normally observed with the CDE treatment, while IFX exhibited minimal effects (Fig. 2B). A similar inhibition on the total-bilirubin level was observed in mice receiving DEX, while in IFX-treated mice the bilurubin levels were unchanged (Fig. 2B). Both DEX and IFX decreased ALP levels (not significantly), indicative of less cholangiocytic injury. This suggests that only DEX injections significantly ameliorate the direct injury on hepatocytes induced by the CDE diet, but both DEX and IFX seem to affect the indirect injury on cholangiocytes.
Strikingly, both DEX and IFX blunted the DR when stained for CK19 (Fig. 2C). Whereas in CDE-treated livers CK19+ cells accumulated both in PV and interlobular zones, in DEX conditions, CK19+ cells were in the PV zone with fewer in its vicinity. On the contrary, IFX co-treatment disturbed CK19+ cell distribution, with CK19+ cells found predominantly in the PV zone (Fig. 2C). In the presence of DEX, the CK19+ cells are smaller in shape, elongated, and with a slight decrease of CK19 immunoreactivity present mainly as duct-like structures or individually but not in clusters as in control CDE livers (Fig. 1B). The overall presence of F4/80+ cells in the hepatic tissue was drastically inhibited by DEX but to a lesser extent by the use of IFX (Fig. 3). Earlier studies reported similar observations when using Clodronate-containing liposome to deplete the KCs19. While in CDE-injured livers, the CK19+ cell expansion was constantly associated with matrix deposition, both DEX and IFX co-treatment repressed laminin and collagen expression and deposition (Fig. 3, hydroxyproline Fig. 2B).

DEX and IFX attenuate the CDE-induced ductular reaction.
(A) Liver sections of control animals and mice that were subjected to a 14 day CDE-diet in co-treatment with either IFX or DEX were stained with CK19, Laminin, Sirius red, and F4/80 antibodies. The pictures display representative photomicrograph. All images were taken in 20x original magnification. (B) Morphometric quantification. All data are mean (+/−SEM) for n = 5/group. *p < 0.05 and ***p < 0.001 difference versus CTL. ###p < 0.001 difference versus treated groups (CDE). £p < 0.05 and £££p < 0.001 difference versus co-treated groups (+/−DEX or +/−IFX). All images were taken in 20x original magnification.
Impact of IFX and DEX on DDC-induced ductular reaction
Upon DDC treatment, the liver colour was not modified by DEX or IFX (Fig. S6) neither was the texture. However, higher levels of ALT indicated that the hepatic injury is two-fold more severe in the DDC/DEX-co-treated mice than in mice treated only with DDC diet, while co-administration of IFX has no impact on ALT levels (Fig. 4B). Interestingly, while both IFX and DEX do not affect the accumulation of porphyrin in DDC-fed livers, only DEX co-administration reduces the levels of bilirubin in serum of co-treated animals. DDC treatment leads to a mild increase in ALP which is not affected by co-treatment of DEX or IFX (Fig. 4B). Co-treatments with DEX or IFX reduce the ductular response as evidenced by CK19 stainings (Fig. 4C); DEX co-treatment results in significantly altered architecture of the biliary system, impeding the pseudo-tubular structure, while IFX did not have this effect. The attenuation of CK19-positivity was also associated with a drastic inhibition of ECM deposition (Fig. 5 and hydroxyproline Fig. 4B). As expected, DEX affected the amount of F4/80+ cells while IFX seems to affect the distribution of the F4/80+ but not the amount. Morphometric and mRNA analyses confirmed these observations (Figs 5B and S7). Altogether, our data show that DEX and IFX reduce ECM deposition and attenuate the DR.

Impact of IFX and DEX on DDC diet mediated liver injury parameters and CK19+ cell distribution.
(A) Experimental design: IFX was injected i.p daily from day 7 of CDE treatment whereas DEX was injected from day 9 onwards in a 14 day time course. (B) Quantification of ALT, ALP, and total-bilirubin in sera and hydroxyproline levels in livers from DDC-treated mice +/− DEX or IFX. (C) Representative immunohistochemistry images for CK19+ cells in DDC-treated livers with or without DEX or IFX are shown. The dotted windows in the left panels are magnified and presented in the middle panels. Higher magnifications of CK19+ cells are given in the two right panels. All data are mean (+/−SEM) for n = 5/group. ***p < 0.001 difference versus CTL, ###p < 0.001 difference versus treated groups (DDC). £££p < 0.001 difference versus co-treated groups (+/−DEX or +/−IFX).

DEX and IFX attenuate the DDC-induced DR.
(A) Liver sections of control animals and mice that were subjected to a 14 day DDC-diet in co-treatment with either IFX or DEX were stained with CK19, Laminin, Sirius red, and F4/80 antibodies. (B) Morphometric quantification. All data are mean (+/−SEM) for n = 5/group. ***p < 0.001 difference versus CTL. ###p < 0.001 difference versus treated groups (DDC). ££p < 0.01, and £££p < 0.001 difference versus co-treated groups (+/−DEX or +/−IFX). All images were taken in 20x original magnification.
IFX and DEX differentially affect the spatial distribution of F4/80+ cells upon CDE and DDC injury
Spatial distribution of F4/80-positive cells in livers damaged by CDE and DDC diets.
Since the difference in amount and distribution of F4/80+ was striking when comparing CDE and DDC diets, we further investigated their localization and quantity in the different dietary setting. In unchallenged livers, single F4/80+ cells with elongated cytoplasm and small nuclei were generally located in the periportal and midlobular regions, sparing the centrilobular regions (Fig. S8). After 7 days of CDE, KCs appeared rounder and enlarged, and were arranged in clusters around the bile ducts, but the majority was found farther away from the portal area. By day 14, F4/80+ cells had formed interconnected networks, which were equally concentrated around the centrilobular and portal veins (Fig. S8A). When quantified, this shift in F4/80+ cell distribution between the CV and PV is even more striking (Fig. S8B). In contrary, upon DDC treatment, F4/80+ cell accumulation was slower, less extensive, and almost exclusively located within the portal fields or in close proximity to porphyrin crystals (Fig. S8).
Impact of DEX and IFX on the spatial distribution of F4/80-positive cells in mice treated with CDE and DDC diets
Analysis of the F4/80+ cell distribution in both models of liver injury, with or without DEX or IFX, showed that F4/80+ cells normally gather massively around the portal vein after 14 days of CDE diet, while such distribution was virtually absent in DEX-administered mice and more comparable to unchallenged livers (Fig. 6A). While F4/80+ cells were confined to midlobular regions around the PV in IFX-administered mice, we also noticed that these cells highly accumulated in CV areas compared to the DEX-administrated mice. Upon cholestatic damage, the F4/80+ cell distribution in co-treated livers by IFX seemed to be similar to that of DDC treatment alone, while DEX exhibits a drastic inhibitory effect on the KCs (Fig. 6B). To gain some mechanistic insight, we investigated the impact of these two treatments on the expression of genes involved in marcrophage recruitment and BEC proliferation (or differentiation) in the DDC- and CDE-treated mice; mRNA levels of the pro-inflammatory cytokines Tnf-α and Il6 and the monocyte chemoattractant protein-1 (Mcp1) (Fig. S9) were decreased by DEX and IFX independently of the liver injury model. Strinkingly, compared to the CDE model, only very low levels of Mcp-1 mRNA levels were detected in the DDC-diet conditions. In contrast, mRNA levels of Tnfr-1 and -2, Jag-1 Notch-1 and -2, IFNɣ and Tweak (Tumour necrosis factor-like weak inducer of apoptosis) were only influenced by DEX. This confirms the more selective action of IFX and suggests that at least in the CDE diet, down regulation of Mcp-1 could be responsible for the reduced amount of F4/80+ cells in DEX and IFX-treated animals.

DEX and IFX modulate the recruitment of F4/80+ cells in both CDE- and DDC- models.
(A) Healthy mice or subjected to a period of 14 day CDE-and DDC-diet in presence or absence of DEX or IFX were analyzed for spatial distribution of F4/80+ cells. Livers sections were stained with F4/80 antibody and counterstained with hematoxylin. The images display representative photomicrograph from portal- and central vein areas. All images were taken in 20x original magnification. (B) Morphometric quantification of stainings performed in (A). All images were taken in 20x original magnification.
DEX and IFX attenuate the adaptive remodeling of the biliary architecture and modulate the proliferative status of ductular cells
While DEX and IFX blunted the increase in CK19+ cells, a subtle difference was visible on the remodeling of the biliary architecture by these 2 drugs (Figs 3A and 5A). To investigate this further, we quantified the distribution of the CK19+-area close to the portal vein and to its vicinity upon CDE treatment in presence or absence of DEX or IFX (see Fig. S10). Notably, while IFX has a stronger inhibitory effect on the CK19+-BEC expansion found in the PV’s vicinity, DEX inhibits more drastically the expansion of the CK19+ cells from the PV’s vicinity (Fig. S10). Our results confirm earlier data showing that in response to different injuries the plasticity of the biliary tree is remodelled accordingly20,21, and suggest that the use of IFX and DEX may have an unprecedented impact on the flexibility of this hepatobiliary system. Contrarily to CDE, in DDC settings the remodeling of the biliary architecture was obvious and was therefore not quantified.
Finally, we hypothesized that at least part of the observed effects of IFX and DEX could be due to an inhibition of BEC, HSC and F4/80+ cell proliferation under these experimental conditions. Overall, both dietary regimens predominantly resulted in an increase in proliferating Ki-67+/Desmin+-HSCs (Fig. S11) and Ki-67+/CK19+-BECs (Fig. 7). Very little proliferation was observed for hepatocytes, LSECs and Kupffer cells (Fig. S11). While DEX decreased the number of Ki-67+ BECs by 63% in CDE- and by 40% in DDC-treated animals, IFX treatment inhibited the amount of Ki-67+ BECs by 27% in CDE- and by 70% in DDC-treated animals. At the same time, DEX and IFX treatment did not significantly affect the number of proliferating HSCs, suggesting that the major impact on fibrogenesis by these two treatments is not due to inhibition of HSC proliferation (Fig. S11).

DEX and IFX influence the proliferative status of the DR in CDE- and DDC-treated livers.
(A) Mice received CDE or DDC dietary regimes for 14 days to induce liver injury. Representative pictures of the detection of CK19/Ki-67 double positive cells in presence or absence of DEX or IFX are shown and (B) Quantification of the amount of Ki-67 positive cells per hepatic cell type per field: hepatocytes (HEP, Albumin+), HSC (Desmin+); LSEC (Lyve1+), KC (F4/80+), BEC (CK19+). Right graph: representation of only Ki-67/CK19 double positive cells per field. PV = portal vein. All data are mean for n = 5/group. ***p < 0.001 difference versus CTL. ###p < 0.001 difference versus treated groups (DDC or CDE). £££p < 0.001 difference versus co-treated groups (+/−DEX or +/−IFX). All images were taken at 20x original magnification.
Discussion
In this study, we evaluated whether targeting the TNF-α axis may be useful in ameliorating liver injury parameters and the DR in the context of a steatotic and a cholestatic model of liver disease. Our results clearly show that (i) TNF-α is essential for the adequate development of the DR in two liver injury models; (ii) in the steatotic CDE model, only DEX is able to reduce the liver injury, while in the cholestatic model DEX worsens the injury; (iii) in the CDE model, the migration of CK19+ cells and the PV localization of F4/80+ cells is dependent on TNF-α. We conclude that both DEX and IFX can modulate the DR and inhibit extracellular matrix deposition in a cholestatic and a steatotic mouse model of liver injury. Overall, our findings are encouraging and indicate that IFX treatment, at least in these steatotic and cholestatic mouse models, is the safer approach since it does not increase liver injury, allows inflammation to take place and ameliorates hepatic proliferation, but efficiently inhibits the DR and extracellular matrix deposition.
Previous studies have shown that drastic modulation of the inflammatory response is possible by depleting macrophages from the organ of interest by means of nanoparticles, like clodronate-containing liposomes22. However, KCs have a biphasic role in liver injury8,23,24 and depleting them at different stages of the DR may make it difficult to predict the outcome. We opted for the systematic administration of DEX. A drawback of this approach is the systemic immunosuppression which makes it also technically difficult to determine which cells have been targeted. Recent studies using models of acute and chronic experimental injury have shown that DEX-loaded liposomes compared to free DEX may be a more efficient tool not only to target macrophages but also to modulate their inflammatory cytokine responses and their migratory properties25,26. In our experiments, DEX did affect ALT levels in DDC-treated mice, nonetheless it still dimished bilirubin levels and decreased collagen and laminin deposition and CK19+ cell expansions. This could mean that DEX encapsulation could reduce the effect on cells that are otherwise sensitive to free DEX, which then could reduce its positive benefit on fibrosis and CK19 expansion25.
After i.p. injection, IFX and DEX enter in the peritoneum, are taken up by the mesenteric drainage system, and then get into the systemic circulation mostly through the hepatic portal system. We consent that our results, although significant in describing the role of inflammatory cells in these models, do not exclude whether those effects are linked to collateral effects, and more particularly on the gut microbiota. Since TNF-α is an important tight-junction regulator of the intestine barrier, injecting DEX or IFX i.p. could also impede the cell signaling at this place. Further studies are thus required to explore whether there is less dysbiosis in co-treated animals.
Lately, substantial evidence demonstrates that sensitization to biological drugs is an important clinical problem. The emergence of “nonresponse,” “loss of response,” or “adverse events” to a certain therapy complicates the management of patients in remission. Particularly with TNF-α agents, loss of response to therapy has been associated to the presence of antibodies that bind to IFX itself, impairing its function and therefore influencing the IFX therapeutic efficacy27. Evidence shows that higher IFX concentrations are associated with greater clinical efficacy in patients with Crohn’s disease and that both presences of antibodies to IFX and low IFX concentrations are associated with worse clinical outcomes28. Consequently, to make a definitive statement on the safety of IFX in our experimental settings, future research should evaluate the optimal IFX dosing. We noticed in our preliminary experiments that dosage and frequency of IFX treatment has a big impact on the outcome; for example 1, 2, or 3 i.p injections of IFX at the beginning or spread over the last week of the liver injury were inadequate to observe a decline of the DR (not shown). However, even with daily injection of IFX, no mortality was seen in the concerned animal groups regardless of the nature of the injury, illustrating that the safety profile of IFX is acceptable. Nevertheless, to better judge potential toxicity of DEX and IFX in future studies, other organs should be investigated as well.
Another aspect we did not consider in this study, is the use of other TNF-α-blocking agents such as adalimumab, golimumab17 or etanercept29 to determine whether one of these clinically approved monoclonal antibodies has a better efficiency and efficacy to abolish the DR and fibrosis. All these TNF-α blockers contain the IgG1 Fc portion, which on one hand improves the in vivo half-life, but on the other hand leads to unwanted effector-mediated cytotoxic effects. Recently, a new anti-TNF-α agent named TNFR-hyFc (TNFR with a hybrid Fc) showed a higher neutralizing activity and even a better protective effect in an arthritis model compared to Etanercept administration30. Comparison of these blockers in different liver injury models would determine if our first choice on IFX was a good one or whether blockers like TNFR-hyFc are more adequate to attenuate the DR from one particular liver disease. Given their susceptibility to physical and chemical degradation processes, anti-TNF-α agents would require the development of stable formulations and specific delivery strategies should be developed to reduce the risk of side effects caused by prolonged presence of anti-TNF-α blockers31. In this study, we only exposed mice for a short term to DEX and IFX, which already resulted in a strongly reduced DR. We do not exclude that longer treatment with these agents in a chronically diseased liver that might need the regenerative capacity of the DR for hepatocyte repopulation could be harmful. However, the only mouse model in which CK19+ cells massively contributed to liver regeneration is the recently reported AhCre+ Mdm2flox/flox mouse, in which Mdm2 inactivation leads to complete senescence of hepatocytes and the activation of CK19+ HSPCs is indeed necessary for survival and complete functional liver reconstitution32. In the used DDC and CDE models, no or only a minor contribution of HSPCs to liver reconstitution could be demonstrated33,34, thereby miminizing the risk of an adverse affect of long-term DR inhibition in these models.
We gained some insight into the mechanisms by which DEX or IFX influence the DR and fibrogenesis in these two liver injury models. Generally, when compared to IFX, DEX administration influences many more signaling pathways as shown by the inhibition of Jag1, Notch1 and -2, as well as Tnfα and Tnfr1 and -2 mRNA expression which were not, or much less, affected by IFX. Both treatments resulted in the downregulation of Mcp1 and Il6 in the CDE setting (Fig. S9), which coincided with less recruitment of F4/80+ cells. This was not the case for the DDC diet, in which only DEX resulted in the lesser expression of Mcp1, while IFX did not influence Mcp1 mRNA levels and did not have an impact on the amount of F4/80+ cells in the DDC-treated livers. However, the levels of Mcp1 mRNA were not increased by DDC while CDE treatment results in a 50 fold increase of Mcp1 mRNA levels, suggesting that probably in DDC-treated mice Mcp1 is not responsible for the macrophage infiltration. Neither IFX, nor DEX, affected the proliferation of F4/80+ cells suggesting that the difference in the F4/80+ cells is mainly due to a difference in recruitment of macrophages. A possible mediator in the effect of DEX on these injury models is Tweak, a known mitogen for HSPCs that acts through the FN14 receptor35 and is necessary for liver fibrosis following chronic CCl4 injury36. Tweak mRNA levels are decreased by DEX in both injury models, which could explain the lesser DR and fibrogenesis when DEX is used (Fig. S9). Of note, recent in vitro experiments with HSCs showed that Tweak could affect proliferation and not activation of HSCs36 while in our experiments HSC is not affected even though DEX decreased Tweak mRNA levels. Moreover, the DEX mediated inhibition of these signaling pathways apparently outperforms the potential downstream negative effects of an increased hepatocytic injury (higher ALT values) in the co-treated DEX/DDC animals (Figs 4 and 5). Thus, mechanistically we can only state that, DEX affects many signaling pathways including Notch, Tnfα and Tweak signaling while IFX is less disruptive, but also leads to a similar diminishment of DR and fibrosis. IFX and DEX both lead to less macrophage recruitment as observed in the CDE diet and perhaps directly affect survival of HSCs by inhibition of TNFα signaling37; IFX directly by acting on TNFα, and both DEX and IFX by downregulation of Tnfα mRNA levels (Fig. S9). Clearly, more studies using cell type specific knock-out mice of these signaling mediators are needed to unravel the exact mechanisms involved in the development of the DR and fibrogensis in these two mouse models of liver injury and to further explain the impact DEX and IFX have on liver disease progression.
Targeting the DR as an approach to improve regeneration or at least inhibit fibrogenesis is not novel. Knight et al. showed, that at least in mice the DR may drive fibrogenesis during chronic liver injury9,38. A recent report demonstrated that cues that regulate the DR also influence fibrosis progression; blocking TWEAK caused a reduction in both HSPC numbers and collagen deposition in a partial hepatectomy mouse model of fibrotic livers39. Studies on NAFLD patients and in vitro studies with BECs and HSCs suggested that CK19+ ductules can influence the progressive portal and bridging fibrosis in NAFLD40. Finally, there is an important DR in advanced alcoholic liver disease patients41,42 of which the extent is correlated with the severity of the disease43,44. Based on our data in both models of liver disease, one could suggest that in alcoholic liver disease patients with important DR, perhaps IFX treatment could have beneficial effects. In fact, clinical trials with alcoholic hepatitis patients treated with IFX alone45,46 or in combination with prednisone47 were promising. Unfortunately, the results of two large randomized controlled trials testing IFX48 or etanercept (soluble TNF receptor)49 were disappointing and reported high rates of infection, perhaps due to the sequential infusions, the major difference with the succesful single dose IFX only trials45,46. Currently, there are no large ongoing studies of targeted treatment for alcoholic hepatitis based on IFX50. Nonetheless, we believe that our findings could be of importance for the further development of drugs for multiple chronic liver disorders that frequently display a ductular response such as patients with alcoholic liver disease or sclerosing cholangitis.
Material and Methods
Animals and Study design
All methods, experimental protocols and animal experiments were performed according to the to European Guidelines for the Care and Use of Laboratory Animals and were approved by the Ethical Committee of Animal Experimentation of the Vrije Universiteit Brussel (VUB, Belgium: Project # 09-13-212-1). Male C57Bl/6J mice (Charles River Laboratories, France) were maintained at temperature, humidity and light-dark cycle (with a day light period from 07.00 a.m. to 19.00 p.m.) controlled conditions in all experiments. Five mice of 4- and 7-week-old of age were allocated per cage and allowed food and water ad libitum. After 1 week of acclimatization, mice were randomly assigned to different experimental groups. In one group, 8-week-old mice were fed with a 3, 5-diethioxycarbonyl-1,4-dihydrocollidine (0, 1% wt/wt) (DDC, C14H21NO4, Sigma-Aldrich, 137030, Bornem, Belgium) containing diet (C1000, Altromin, Lage, Germany) for 7 and 14 days to induce a cholangiocytic injury51. In another group, 5-week-old mice were fed with a diet deficient in choline (MP Biomedical, Irvine, USA) supplemented with 0, 15% (wt/vol) ethionine (Sigma-Aldrich, E5139) in drinking water (CDE) for 7 and 14 days to induce a hepatocyte injury52. Untreated, age-, and sex-matched mice were used as control. Food and water were changed every 3 days and ethionine-containing water bottles were protected from light.
In a first set of experiments, mice were treated with dexamethasone (DEX, 1mg/kg, Sigma) by a daily i.p. injection during the last 5 consecutive days of 2 weeks DDC or CDE-treatment. In a second set of experiments, after 1 week of dietary treatment, the mice were i.p.-injected daily with Infliximab (IFX or RemicadeTM, MSD, Belgium, 25μg/g) for an additional 7 days after the beginning of the treatment. IFX and DEX were diluted in sterile PBS and then filtered through 0.22 μm filter prior to injection. Control group animals received saline injections. Regardless of the treatment, males with same age by group were used and 100 μL maximum of solution was injected.
See Supplementary Information for other standard protocols, materials and products used in this study.
Additional Information
How to cite this article: Verhulst, S. et al. Infliximab and Dexamethasone Attenuate the Ductular Reaction in Mice. Sci. Rep. 6, 36586; doi: 10.1038/srep36586 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Itoh, T. & Miyajima, A. Liver regeneration by stem/progenitor cells. Hepatology 59, 1617–1626 (2014).
Williams, M. J., Clouston, A. D. & Forbes, S. J. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 146, 349–356 (2014).
Richardson, M. M. et al. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology 133, 80–90 (2007).
Viebahn, C. S. et al. Invading macrophages play a major role in the liver progenitor cell response to chronic liver injury. J Hepatol 53, 500–507 (2010).
Theise, N. D. et al. The canals of Hering and hepatic stem cells in humans. Hepatology 30, 1425–1433 (1999).
Miyajima, A., Tanaka, M. & Itoh, T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 14, 561–574 (2014).
Murray, P. J. & Wynn, T. A. Protective and pathogenic functions of macrophage subsets. Nature reviews. Immunology 11, 723–737 (2011).
Chazaud, B. Macrophages: supportive cells for tissue repair and regeneration. Immunobiology 219, 172–178 (2014).
Knight, B. et al. Attenuated liver progenitor (oval) cell and fibrogenic responses to the choline deficient, ethionine supplemented diet in the BALB/c inbred strain of mice. Journal of Hepatology 46, 134–141 (2007).
Boulter, L. et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med 18, 572–579 (2012).
Bird, T. G. et al. Manipulation of liver regeneration with macrophages to influence the hepatic progenitor cell niche. The Lancet 381, S23 (2013).
Elsegood, C. L. et al. Kupffer cell-monocyte communication is essential for initiating murine liver progenitor cell-mediated liver regeneration. Hepatology 62, 1272–1284 (2015).
Nestorov, I. Clinical pharmacokinetics of TNF antagonists: how do they differ? Semin Arthritis Rheum 34, 12–18 (2005).
Bradley, J. R. TNF-mediated inflammatory disease. J Pathol 214, 149–160 (2008).
Karampetsou, M. P., Liossis, S. N. & Sfikakis, P. P. TNF-alpha antagonists beyond approved indications: stories of success and prospects for the future. QJM 103, 917–928 (2010).
Waters, J. P., Pober, J. S. & Bradley, J. R. Tumour necrosis factor in infectious disease. J Pathol 230, 132–147 (2013).
Kaymakcalan, Z. et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol 131, 308–316 (2009).
Barnes, P. J. Corticosteroids: the drugs to beat. Eur J Pharmacol 533, 2–14 (2006).
Van Hul, N. et al. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am J Pathol 179, 1839–1850 (2011).
Vartak, N. et al. Cholestasis-induced adaptive remodeling of interlobular bile ducts. Hepatology 63, 951–964 (2015).
Kaneko, K., Kamimoto, K., Miyajima, A. & Itoh, T. Adaptive remodeling of the biliary architecture underlies liver homeostasis. Hepatology 61, 2056–2066 (2015).
Van Rooijen, N. & Sanders, A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. Journal of immunological methods 174, 83–93 (1994).
Duffield, J. S. et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. Journal of Clinical Investigation 115, 56–65 (2005).
Tacke, F. & Zimmermann, H. W. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol 60, 1090–1096 (2014).
Bartneck, M. et al. Liposomal encapsulation of dexamethasone modulates cytotoxicity, inflammatory cytokine response, and migratory properties of primary human macrophages. Nanomedicine 10, 1209–1220 (2014).
Bartneck, M. et al. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials 37, 367–382 (2015).
Baert, F. et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med 348, 601–608 (2003).
Vande Casteele, N. et al. The relationship between infliximab concentrations, antibodies to infliximab and disease activity in Crohn’s disease. Gut 64, 1539–1545 (2014).
Viswanathan, P., Kapoor, S., Kumaran, V., Joseph, B. & Gupta, S. Etanercept blocks inflammatory responses orchestrated by TNF-alpha to promote transplanted cell engraftment and proliferation in rat liver. Hepatology 60, 1378–1388 (2014).
Lee, J. H. et al. The pharmacology study of a new recombinant TNF receptor-hyFc fusion protein. Biologicals 41, 77–83 (2013).
Tosello-Trampont, A. C., Landes, S. G., Nguyen, V., Novobrantseva, T. I. & Hahn, Y. S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J Biol Chem 287, 40161–40172 (2012).
Lu, W. Y. et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol 17, 971–983 (2015).
Rodrigo-Torres, D. et al. The biliary epithelium gives rise to liver progenitor cells. Hepatology 60, 1367–1377 (2014).
Espanol-Suner, R. et al. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 143, 1564–1575 e1567 (2012).
Jakubowski, A. et al. TWEAK induces liver progenitor cell proliferation. J Clin Invest 115, 2330–2340 (2005).
Wilhelm, A. et al. Interaction of TWEAK with Fn14 leads to the progression of fibrotic liver disease by directly modulating hepatic stellate cell proliferation. J Pathol 239, 109–121 (2016).
Pradere, J. P. et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473 (2013).
Knight, B., Lim, R., Yeoh, G. C. & Olynyk, J. K. Interferon-gamma exacerbates liver damage, the hepatic progenitor cell response and fibrosis in a mouse model of chronic liver injury. J Hepatol 47, 826–833 (2007).
Kuramitsu, K. et al. Failure of fibrotic liver regeneration in mice is linked to a severe fibrogenic response driven by hepatic progenitor cell activation. Am J Pathol 183, 182–194 (2013).
Chiba, M. et al. Participation of bile ductular cells in the pathological progression of non-alcoholic fatty liver disease. Journal of clinical pathology 64, 564–570 (2011).
Roskams, T. et al. Oxidative Stress and Oval Cell Accumulation in Mice and Humans with Alcoholic and Nonalcoholic Fatty Liver Disease. The American Journal of Pathology 163, 1301–1311 (2003).
Jung, Y. et al. Accumulation of hedgehog-responsive progenitors parallels alcoholic liver disease severity in mice and humans. Gastroenterology 134, 1532–1543 (2008).
Lowes, K. N., Brennan, B. A., Yeoh, G. C. & Olynyk, J. K. Oval Cell Numbers in Human Chronic Liver Diseases Are Directly Related to Disease Severity. The American Journal of Pathology 154, 537–541 (1999).
Sancho-Bru, P. et al. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology 55, 1931–1941 (2012).
Tilg, H. et al. Anti-tumor necrosis factor-alpha monoclonal antibody therapy in severe alcoholic hepatitis. Journal of Hepatology 38, 419–425 (2003).
Sharma, P., Kumar, A., Sharma, B. C. & Sarin, S. K. Infliximab monotherapy for severe alcoholic hepatitis and predictors of survival: An open label trial. Journal of Hepatology 50, 584–591 (2009).
Spahr, L. et al. Combination of steroids with infliximab or placebo in severe alcoholic hepatitis: a randomized controlled pilot study. Journal of Hepatology 37, 448–455 (2002).
Naveau, S. et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 39, 1390–1397 (2004).
Boetticher, N. C. et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology 135, 1953–1960 (2008).
Louvet, A. & Mathurin, P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol 12, 231–242 (2015).
Fickert, P. et al. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am J Pathol 171, 525–536 (2007).
Akhurst, B. et al. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology 34, 519–522 (2001).
Acknowledgements
We thank D. Blyweert and K. Lagaisse for their excellent technical assistance. The TROMA-III antibody was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of NICHD and maintained by The University of Iowa. We are grateful to Prof. P. In ‘t Veld for the use of the Nikon microscope. S.V. was supported by the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT/SB/121548), W.K.S.: Foundation for Liver Research, UK; K.H.: Research Foundation Flanders (FWO; G.0428.09N); VUB- research council-OZR1914; L.D., J.B. and L.v.G.: Interuniversity Attraction Poles (Federal Science Policy–BELSPO P7/47); Innoviris-Impulse program 2011-IP-LS-104–Brustem (Brussels Government) and FWO G.0348.13N.
Author information
Authors and Affiliations
Contributions
S.V., L.D., K.H. and L.v.G. participated in the design of this study, performed data collection analysis, and drafted the manuscript; S.V., L.D. and H.R. participated in functional validation experiments; S.W., J.B. and A.C. helped with the interpretation of the data and revision of the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Verhulst, S., Best, J., Syn, WK. et al. Infliximab and Dexamethasone Attenuate the Ductular Reaction in Mice. Sci Rep 6, 36586 (2016). https://doi.org/10.1038/srep36586
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36586
This article is cited by
-
TNF in the liver: targeting a central player in inflammation
Seminars in Immunopathology (2022)
-
RNA-sequencing-based comparative analysis of human hepatic progenitor cells and their niche from alcoholic steatohepatitis livers
Cell Death & Disease (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
