
Abstract
LIM-homeodomain (HD) transcription factors form a multimeric complex and assign neuronal subtype identities, as demonstrated by the hexameric ISL1-LHX3 complex which gives rise to somatic motor (SM) neurons. However, the roles of combinatorial LIM code in motor neuron diversification and their subsequent differentiation is much less well understood. In the present study, we demonstrate that the ISL1 controls postmitotic cranial branchiomotor (BM) neurons including the positioning of the cell bodies and peripheral axon pathfinding. Unlike SM neurons, which transform into interneurons, BM neurons are normal in number and in marker expression in Isl1 mutant mice. Nevertheless, the movement of trigeminal and facial BM somata is stalled, and their peripheral axons are fewer or misrouted, with ectopic branches. Among genes whose expression level changes in previous ChIP-seq and microarray analyses in Isl1-deficient cell lines, we found that Slit2 transcript was almost absent from BM neurons of Isl1 mutants. Both ISL1-LHX3 and ISL1-LHX4 bound to the Slit2 enhancer and drove endogenous Slit2 expression in SM and BM neurons. Our findings suggest that combinations of ISL1 and LHX factors establish cell-type specificity and functional diversity in terms of motor neuron identities and/or axon development.
Similar content being viewed by others

Introduction
Motor neurons (MNs) transmit signals from the CNS to peripheral muscles to control voluntary and involuntary movements. For instance, cranial motor neurons, which control head and neck movements comprise three subtypes based on their functions and origins: branchiomotor, visceral motor (VM) and somatic motor neurons. BM neurons control the movement of tongue and jaw and facial expression, and VM neurons regulate involuntary movement as part of the autonomic nervous system. SM neurons innervate skeletal muscles and control voluntary movements. The origins and major transcription programs of BM/VM neurons differ from those of SM neurons such as PHOX2 and ISL1 factors for BM/VM neurons and LHX3 and ISL1 for SM neurons1,2,3. However, it is unclear whether the signals for targeting the axons of individual cranial motor neurons to distinct muscle targets are the same or different4,5.
ISLET1 (ISL1) is a member of the LIM-HD transcription factor family present in all MNs. Its role in acquisition of motor neuron identity in the spinal cord is well-established6,7,8,9,10. When ISL1 expression is reduced, SM neurons transdifferentiate into V2a interneuron-like cells in the spinal cord2,8. ISL1 is also expressed in postmitotic MNs, raising the possibility that it plays additional roles. Indeed, several lines of evidence suggested that it may control MN axon pathfinding. In zebrafish, peripheral projections of RB primary sensory neurons and trigeminal neurons are affected when isl1 or its paralogue isl2 is mutated7,9,11. Axon pathfinding and neurotransmitter identity are compromised in some neurons in isl1 null flies12. In mice, when the ISL1 level is reduced, peripheral projections of retinal axons as well as motor and sensory neurons are disrupted, all of which support the potential roles of ISL1 in axon navigation13,14.
LIM-HD transcription factors build multimeric complex via interaction with LBD1 as demonstrated by previous biochemical, structural and genetic studies2,15,16,17,18. Unlike other LIM-HD family members, ISL1 appears to act together with other LHX factors in the CNS such as spinal cord motor neurons and striatal interneurons2,19. However, it is uncertain whether a similar combinatorial LIM-code is employed in most neurons in general. For instance, BM neurons in the hindbrain also express ISL1 but no LHX factors that interact with ISL1 are known. Furthermore, diverse combinatorial LIM-codes may control multiple biological functions in the same or different cells.
To investigate the postmitotic roles of ISL1-based LIM-complex in axon guidance, we focused on cranial BM neurons, which retain their identity and project axons to the periphery in the absence of ISL18. We found that, in Isl1 compound mutant mice, many subpopulations of cranial motor axons were defasciculated or misrouted. Notably, misprojection of BM axons accompanied arrest of the movement of BM somata, indicating that both peripheral and central mechanisms were affected in the absence of ISL1. Analyzing previous Chip-seq and microarray experiments and in situ hybridization of candidate genes revealed that Slit2 transcription was controlled by ISL1, with the help of LHX4, and that Slit2 mRNA levels were downregulated in Isl1-deficient BM neurons. We therefore suggest that Slit2 is a downstream target of ISL1 in cranial motor neurons and ISL1 controls axon pathfinding in cranial motor neurons.
Results
Specification of cranial motor neurons in Isl1 compound mutant mice
Isl1 null mutants do not survive beyond E9.5 due to cardiovascular defects, which make it impossible to investigate their neural development10. We instead used Isl1 compound mice (Isl1hypo/KO) carrying one Isl1 hypomorphic allele and one Isl1 null allele8,10,14. A significant reduction in ISL1 immunoreactivity in the spinal cord was observed in similar Isl1 mutant mice with an Isl1 hypo allele and an Isl1nLacZ knock-in allele14. Our Isl1 compound mice survived until E11.75, which allowed to examine the roles of ISL1 in motor neurons. We also used Isl1 conditional knockout (cKO) mice with a CNS-specific Nestin-Cre that efficiently removed ISL1 protein in the hindbrain8. In wild-type E11.5 hindbrain flat-mount tissues and transverse sections, ISL1 is present in all cranial motor neurons including the facial branchiomotor (FBM) neurons, which migrate from r4 to r6 (Fig. 1A,E). Trigeminal (V) in r2 and FBM (VII) neurons in r4 to r6 co-expressed PHOX2B and ISL1 (Fig. 1I,L,O,R)3,4. In the Isl1 compound mutant mice, ISL1 immunoreactivity was reduced by 40% in r4 FBMs and undetectable in caudal hindbrains (r7–8) (Fig. 1C,G,H,DD,EE). To trace cranial motor neurons when the ISL1 level was low, we crossed Isl1 compound mutant mice with ISLMN: GFP-F transgenic mice in which all motor neurons are GFP-labeled20. Flat-mounted hindbrains of E11.5 littermate control mice showed all cranial motor somata and axons labeled by GFP (Fig. 1B). In flat-mounted hindbrains of the Isl1 compound mutants, FBM neurons were present in r4 and r5 and a few SM somata were in the caudal hindbrain (Fig. 1D). We also labeled Isl1 mutant neurons with a delta-Isl1 riboprobe designed to detect partial non-functional transcripts that overlap with Tbx20 transcripts, which would indicate that ISL1-low cells survive and express BM neuronal markers (Supplementary Figure S1A–R). Mutant BM neurons labeled with PHOX2B and ISL1/2 were comparable in number with those in their littermate control in the r2 trigeminal nerve, while greater numbers of r4 FBM neurons were dispersed laterally in the Isl1 compound and cKO mutants (Fig. 1I–N). Overall, the generation and initial specification of the trigeminal and FBM neurons appeared relatively normal in Isl1 mutant mice.

Specification of cranial motor neurons in Isl1hypo/KO mice.
(A,C,E–H) Immunostaining of ISL1 in flat-mounted preparations and transverse sections of E11.5 hindbrains. ISL1 immunoreactivity is reduced in r4 (compare arrows in A,E,C,G) and absent in r7–8 (H). (B,D) Flat-mounted preparation of E11.5 ISLMN: GFP-F hindbrains. SM neurons are missing from the caudal brain in the Isl1 compound mutant mice (compare brackets in B,D). (I–CC,FF,GG) Immunostaining and quantification of the BMN marker PHOX2B, the SMN marker HB9, V2a interneuron marker CHX10 and ISL1/2 in the E11.5 transverse hindbrain sections. In both Isl1 compound and cKO hindbrains, FBM neurons stall at r5 and Hb9+ SM neurons are absent from caudal hindbrains, in which more CHX10+ V2a interneurons arise. Note that the r2 trigeminal nuclei are smaller (arrows, J,K) and some r4 FBM neurons migrate laterally (arrowheads, N,Q) in the Isl1 mutants (FF: n = 6; (GG) n = 6; number of sections). (DD,EE) ISL1 immunofluorescence intensity in r4 and the number of ISL1+ cells in r7–8. n = 45 (DD), n = 18 (EE). Error bars represent s.e.m. *p < 0.05 compared with control, ***p < 0.001 compared with control, unpaired Student’s t-test in (DD,EE,GG), Mann-Whitney rank sum test in (FF). Scale bars: in (D), 250 μm for (A–D) in (H), 100 μm for (E–H); in (CC) 100 μm for (I-CC).
Next we tested whether BM nuclei were correctly positioned after cell body migration: trigeminal somata move from medial to lateral at r2, and FBM somata migrate tangentially from r4 to r621. Trigeminal neurons migrated normally but their nuclei were smaller in Isl1 compound mutant mice and, to a greater degree, in Isl1 cKO mice, indicating that ISL1 is required for lateral migration of trigeminal neurons (Fig. 1I–K). FBM neurons in transverse sections of r4 to r6 adopt characteristic shapes in a medial position in r4 and r5, and a lateral position in r6 (Fig. 1L,O,R). In Isl1 compound and cKO mice, most FBM neurons remained in r4 and r5 and failed to arrive at r6 (r4; control, 225.3 ± 19.2 cells; Isl1hypo/KO, 397.0 ± 16.5 cells, p < 0.001), (r5; control, 182.3 ± 5.1 cells; Isl1hypo/KO, 261.3 ± 16.2 cells, p = 0.029), (r6; control, 147.3 ± 5.9 cells; Isl1hypo/KO, 21.8 ± 13.1 cells, p < 0.001) (Fig. 1M,N,P,Q,S,T,FF). In addition, r4 and r5 FBM neurons tended to spread laterally in Isl1 compound mutant mice, and more so in Isl1 cKO mice (Fig. 1N,Q). Together these observations indicate that FBM neurons arise normally when the ISL1 level is low, but their migration is disrupted.
We examined whether the SM neurons in r5 and the caudal hindbrain are intact in Isl1 compound mutant mice, despite the absence of ISL1immunoreactivity (Fig. 1EE). Previously, elimination of ISL1 in the spinal cord was found to result in an increase of V2a interneurons at the expense of SM neurons8. Similarly, we observed that HB9-expressing motor neurons disappeared (control, 57.5 ± 3.8 cells; Isl1hypo/KO, 0.0 ± 0.0 cells, p < 0.001) and CHX10+ V2a interneurons appeared (control, 95.6 ± 5.0 cells; Isl1hypo/KO, 172.5 ± 4.5 cells, p < 0.001) in r5 and r7–8 (Fig. 1V,W,BB,CC,GG). Thus, the production of SM neurons is disrupted in Isl1 compound mutant mice.
BM axons are defective in Isl1 compound mutant mice
To trace the axonal projections of BM/VM neurons, we examined Isl1 compound mutant mice carrying the ISLMN: GFP-F reporter allele in which BM and SM neurons are labeled with GFP20. Embryos were immunostained for GFP to detect motor neurons. Oculomotor neurons of Isl1 mutants were defasciculated at E10.5, and became relatively normal at E11.5 (Fig. 2A–D). The mandibular branches, the motor part of the trigeminal nerve, form a thick axon bundle growing toward the target muscles with fasciculated axon tips (Fig. 2A,C,E,G). Interestingly, the trigeminal axons of Isl1 compound mutants were defasciculated at distal axons and developed prominent extra branch in the middle of primary axon bundle (primary axon length; control, 1960.9 ± 70.0 μm; Isl1hypo/KO, 1506.2 ± 155.3 μm, p = 0.008) (length of extra branches; control, 0.0 ± 0.0 μm; Isl1hypo/KO, 1031.7 ± 121.5 μm, p < 0.001) (Fig. 2A–H,M,N, Supplementary Figure S2I). The FBM axon bundles of Isl1 mutants were thinner and shorter (axon length; control, 1809.2 ± 85.0 μm; Isl1hypo/KO, 1084.4 ± 104.1 μm, p < 0.001) (axon thickness; control, 105.7 ± 6.5 μm; Isl1hypo/KO, 57.7 ± 6.5 μm, p < 0.001) (Fig. 2A–H,O,P, Supplementary Figure S2I). To trace inner ear efferent (IEE) projection, embryo heads were immunostained as open-book flat-mounts. IEE axons exited from the vestibular nerve root and almost reached the cochlear at E11.5 (Fig. 2Q,R)22. However, IEE axons in Isl1 compound mutants were short, disrupted and hardly extended towards the inner ear (Fig. 2S,T). The number and position of somata labeled with GATA3 within the neural tube were normal in Isl1 mutant embryos, indicating that decreased axon outgrowth in the periphery is not simply due to reduction in their cell number (control, 40.7 ± 2.3 cells; Isl1hypo/hypo, 45.5 ± 4.1 cells) (Supplementary Figure S3C–E)22. And there was no obvious sign of cell death or axon degeneration in FBM and IEE neurons of Isl1 compound mutants since no cleaved-CASPASE-3 immunoreactivity was found in them and their explants showed robust axon outgrowth in vitro (neurite length; control, 1.0 ± 0.1 fold; Isl1hypo/KO, 1.42 ± 0.2 fold, p = 0.016), (neurite number; control, 16.3 ± 1.3 neurites, Isl1hypo/KO, 30.7 ± 2.8 neurites, p < 0.001) (Supplementary Figure S2A–H). The normal exit point of FBM and IEE neurons in the neural tube is at the lateral position of r4 (Fig. 2U). In E11.5 flat-mounted hindbrains of Isl1 compound mutant mice, however, FBM and IEE axons had additional exit points at more medial positions with disorganized projections (Fig. 2V, Supplementary Figure S3B). In summary, axon pathfinding by BM neurons was disrupted when the ISL1 level was reduced.

Axon defects of cranial MNs in Isl1hypo/KO mice.
(A–D) Wholemount GFP immunostaining of E10.5 and E11.5 ISLMN: GFP-F reporter embryos. Oculomotor axons of Isl1hypo/KO mice are defasciculated at E10.5 (arrowhead, B). (E–L) Magnified views of images in (A–D). The trigeminal mandibular (V) nerves of Isl1hypo/KO mice are defasciculated (arrowhead in E,G vs arrowhead in F,H) and develop ectopic branches (open arrowheads, F,H). The facial nerve (VII) are fewer (arrows in E,G vs arrows in F,H) and the hypoglossal nerve (XII) is absent (arrow in I,K, arrow in J,L) (E10.5: n ≥ 3; E11.5: n ≥ 7; number of embryos). (M–P) Quantification of nerve length and thickness of axons. Error bars represent s.e.m. *p < 0.05, ***p < 0.001, unpaired Student’s t-test. (Q–T) Open-book flat-mount preparation of E11.5 hindbrains. IEE projections are short and disorganized in Isl1hypo/KO mice (arrowhead, T) (control: n = 5; Isl1hypo/KO mice: n = 3; number of embryos). (U,V) Flat-mounted preparation of E11.5 hindbrains. Migration of FBM neurons is arrested in r5 and their exit points are disrupted (arrowhead in U vs arrowheads in V) (control: n = 4; Isl1hypo/KO mice: n = 3; number of embryos). (W,X) Whole-mount views of E11.5 Hb9::GFP mice. Axons of hypoglossal (XII) neurons and spinal cord MNs in Isl1hypo/KO mice are severely reduced or missing (arrowheads) (control: n = 4; Isl1hypo/KO mice: n = 3; number of embryos). (Y-BB) Flat-mount views of E11.5 Hb9::GFP hindbrains. The abducens neurons in r5 and SM somata are reduced and the interneuron-like longitudinal projections were found in the Isl1hypo/KO mice (arrows, BB) (control: n = 5; Isl1hypo/KO mice: n = 2; number of embryos). III, oculomotor; (V) trigeminal mandibular; VII, facial nerve; IX, glossopharyngeal; X, vagal; XI, spinal accessory; XII, hypoglossal nerve; FBMf, facial branchio motor neuron fibers; IEEf, inner ear efferent fibers. Scale bars: in (D), 500 μm for (A–D) in (L), 250 μm for (E–L) in (T), 500 μm for (Q–T) in (V), 200 μm for (U,V) in (X), 250 μm for (W,X) in (BB), 200 μm for (Y-BB).
We also examined the axon projections of SM neurons, whose identity was affected in Isl1 compound mutant mice. In these mutants, SM axons such as those of the hypoglossal nerve (XII) were almost absent with only a few aberrant axons to be seen (Fig. 2J,L). Similar results were obtained in Hb9::GFP transgenic mice, in which SM neurons are selectively labeled (Fig. 2W)8,10. Peripheral axons of SM neurons were almost absent from caudal hindbrains, and a few misrouted projections remained in cervical neurons (Fig. 2X). Cell bodies disappeared and interneuron-like trajectories spanning the A-P axis of the hindbrain were visible in flat-mounted hindbrains (Fig. 2Y–BB). Thus, axon projection in SM neurons is also disrupted in Isl1 compound mutant mice.
Slit2 signaling is defective in Isl1 mutant BM neurons
Previously we performed microarray screens of embryonic stem cells (ESCs) derived from Isl1 knockout cells23. Since ISL1 is a transactivator, we focused on 683 genes significantly downregulated in Isl1 null cells (P < 0.05 with Bonferroni correction). To search for direct downstream targets of ISL1, we also re-analyzed previously published ChIP-seq data for ISL1 genomic binding sites retrieved from an ESC line, induced by NGN2, ISL1, PHOX2B (NIP) with BM/VM neuronal properties, and combined it with microarray results from Isl1 null cells1. About 1,590 genes had significant binding peaks (>1.5-fold with P < 0.01) for ISL1 in their vicinity (within ± 2 kb of gene boundaries), and 83 of them were downregulated in Isl1-deficient cells. By adopting additional microarray data obtained from NesE-PHOX2B ESC line derived visceral MNs, we finally selected 13 genes as potential ISL1 targets in BM neurons; these included genes for choline acetyltransferase (Chat), neuropilin1 (Nrp1) and Slit2 (Fig. 3A, Supplementary Table S1)24. ISL1 is expressed in both BM and SM neurons, therefore we investigated whether ISL1-mediated transcriptional regulation is conserved in BM and SM neurons. We analyzed another ChIP-seq and microarray datasets derived from two independent ESC lines with SM characteristics, NIL (NGN2, ISL1, LHX3) and NesE-OLIG2, which differentiated from mESCs to SM neurons by expressing OLIG2 under the Nestin enhancer1,24. As a result, we identified 32 genes as putative downstream genes of ISLl in SM neurons (Fig. 3B, Supplementary Table S1). To gain a better understanding of shared or cell type-specific transcriptional control of ISL1 in BM neurons, we focused on 13 genes altered in BMN-ESCs; 8 of them were altered in both BMN and SMN-ESCs (BM & SM genes) and 5 of them were only altered in BMN-ESCs (BM genes) (Fig. 3C). Thus, transcriptional control of ISL1 may be partly conserved between BM and SM neurons.

Reduced Slit2 transcripts in Isl1 mutant BMNs.
(A,B) Venn diagrams indicating the overlap between ISL1 binding sites in a Chip-seq analysis and two independent microarray analyses in BM (A) and SM (B) cells. The list of genes found in both BM and SM cellular contexts is shown in red. (C) Heatmap of genes associated with BM and SM neurons. Red indicates higher relative expression, and blue indicates lower relative expression compared to the median values of the two groups. (D–Q) At E11.5, Slit2 mRNA is undetectable in oculomotor (E), trigeminal (G), facial (I,K,M) and SM (O) neurons (white arrowheads) unlike their littermate controls (black arrowheads, D,F,H,J,L,N). (control: n = 3; Isl1 mutant: n = 3; number of embryos). Scale bars: in (O), 100 μm for (D–O); in (Q), 200 μm for (P,Q).
To verify whether transcript levels of genes selected by the bioinformatics analysis are actually altered in Isl1-deficient BM neurons, we examined mRNA or protein levels of major axon guidance genes or others including NRP1, Slit2, CHAT, TAG-1 and Unc5c. All of them were present in BM neurons, however, only Slit2 transcript levels were diminished in BM neurons of Isl1 mutants (Fig. 3D–Q, Supplementary Figure S4A–H). Slit2 mRNA levels were high in wild type SM neurons and floor plates, and relatively low but definite in post-migrated cranial motor neurons, including oculomotor neurons and trigeminal and migrating FBM neurons (Fig. 3D,F,H,J,L,N,P)25. Remarkably, the levels of Slit2 transcripts in oculomotor, trigeminal and FBM neurons were greatly attenuated in Isl1 compound mutant mice, whereas Slit2 expression in the floor plate was normal (Fig. 3E,G,I,K,M,O,Q). Slit2 expression in SM neurons also disappeared in Isl1 mutants since SM neurons transfate to become interneurons (Fig. 3O, see Fig. 1BB,CC). Transcripts of the related ligands Slit1 and Slit3, and their receptors Robo1 and Robo2, were not changed in the compound mutants nor selected as candidate genes in the bioinformatics analysis (Supplementary Figure S4I–R).
Since ROBO-SLIT signaling controls axon navigation, the extra branches in trigeminal axons of Isl1 mutants could be due to defective ROBO-SLIT signaling26,27. In line with this, FBM somata were mispositioned when Robo1 and Robo2 are downregulated, which indicates that ROBO-SLIT signaling is important in developing FBM neurons28. We therefore examined the FBM projections in Robo1−/−; Robo2−/− mice traced with the ISLMN: GFP-F reporter29. Trigeminal mandibular (V) axons of Robo mutants were defasciculated or had extra branches (primary trigeminal axon length; control, 944.4 ± 75.4 μm; Robo1−/−; Robo2−/−, 809.9 ± 93.2 μm) (length of extra branches; control, 0.0 ± 0.0 μm; Robo1−/−; Robo2−/−, 398.2 ± 33.2 μm, p < 0.001) (Supplementary Figure S5). In addition, FBM axons were thinner and shorter in Robo mutants, similar to axons of Isl1 mutant mice (facial axon length; control, 1171.7 ± 49.2 μm; Robo1−/−; Robo2−/−, 976.9 ± 56.3 μm, p = 0.046) (facial axon thickness; control, 86.5 ± 5.1 μm; Robo1−/−; Robo2−/−, 45.1 ± 4.9 μm, p = 0.002) (Supplementary Figure S5). This is not due to reduced number of FBM neurons since it was reported that the number of FBM somata in Robo1−/−; Robo2−/− were comparable28. Although we cannot completely exclude the possibility that other genes controlled by Isl1 are involved, projection errors found in Isl1 compound mutant could be affected by defective ROBO-SLIT signaling at least in part.
ISL1 controls Slit2 transcription in SM and BM neurons together with LHX3 and LHX4
ISL1 is a LIM-HD transcription factor, whose N-terminal LIM domains mediate protein-protein interactions. In the cortex and spinal cord, ISL1 forms complexes with LHX8 and LHX3, respectively2,19. We reasoned that it may similarly require a LIM-HD transcription factor in its role in BM neurons. The only LIM-HD transcription factor known to be present in BM neurons is LHX4 (Fig. 4A–J)30. Lhx4 mRNA was found to be present in oculomotor neurons, migrating and postmitotic trigeminal and facial motor neurons, and its expression nicely overlapped with Slit2 transcripts, which made it a plausible candidate for interacting with ISL1 (Fig. 4D–I, and see Fig. 3). If LHX4 did form a complex with ISL1 in BMNs, removing Lhx4 will also downregulate Slit2 expression and may cause axon defects. To test this hypothesis, we examined Slit2 mRNA expression in BM neurons of Lhx4 knock-out mice31. Adjacent sections were used to locate BM neurons labeled with ISL1. In littermate controls, BM neurons were migrating from r4 to r6 and Slit2 transcripts were found in r5 (Supplementary Figure S6A,C). However, BM neurons were located in r4-r6 of the Lhx4 knock-out hindbrain but Slit2 transcripts were undetectable (Supplementary Figure S6B,D). Together these results suggest that ISL1-LHX4 hexamer complexes control Slit2 transcription in BMNs.

LHX4 is present in BMNs.
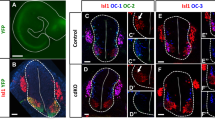
(A–C’) LHX1/2, LHX3 and LHX9 are not detectable in BM neurons labeled in green in transverse sections of E11.5 hindbrains of ISLMN: GFP-F mice (white dotted lines). (D–I) Lhx4 transcript is present in BM neurons of E11.5 transverse hindbrain sections. (J) A list of LIM-homeodomain transcription factors present in BMNs. Scale bars: in (C’), 100 μm for (A–C’) in (I), 100 μm for (D–I).
We searched for ISL1 and LHX binding sites in the genomic locus of Slit2 in the ChIP-seq data in NIL cells and chose the highest ChIP-seq peak, which was located in the 6th intron of Slit2 (hereafter referred to as the Slit2 enhancer) (Fig. 5A)1. Interestingly, ChIP-seq data for LHX3 binding loci also showed the highest peak at the same genomic region (Fig. 5A)32,33,34. This indicates that ISL1 and LHX3 together may drive Slit2 transcription in SM neurons, probably by forming the ISL1 and LHX3 hexameric complex2,35. We found that a GFP reporter carrying the Slit2 enhancer was active in both BM and SM neurons when electroporated into the hindbrains and spinal cords, respectively (Fig. 5B,R). To pinpoint the cells in which the Slit2 enhancer was active in the hindbrain, we generated a nucGFP reporter and introduced it by electroporation together with CMV::mCherry as an internal control. When ISL1 or LHX4 was introduced by electroporation, the nucGFP signal was mostly confined to the FBM nucleus, as in the control group (control, 7.7 ± 1.5 cells; ISL1, 10.9 ± 2.0 cells; LHX4, 11.8 ± 1.6 cells) (Fig. 5C,D,GG). When both ISL1 and LHX4 were introduced, the number of GFP-expressing cells medial to the facial nucleus increased in the hindbrains (ISL1 + LHX4, 152.8 ± 28.1 cells, p < 0.001) (Fig. 5E,GG). More importantly, in this medial region, upregulation of ckSlit2 transcript was observed, indicating that ISL1 and LHX4 not only activate the Slit2 enhancer but can also drive ectopic expression of Slit2 mRNA (arbitrary unit of Slit2 intensity; control, 1.0 ± 0.0 fold; ISL1, 1.0 ± 0.1 fold; LHX4, 0.9 ± 0.4 fold; ISL1 + LHX4, 1.3 ± 0.0 fold, p < 0.001) (Fig. 5Q,KK). This was not due to ectopic production of BM neurons since no additional BM neurons labeled with TBX20 were found in this region (control, 57.8 ± 7.75 cells; ISL1, 58.0 ± 8.0 cells; LHX4, 52.8 ± 5.1 cells; ISL1 + LHX4, 57.8 ± 2.4 cells) (Fig. 5I,M,II). We observed similar effects with ISL1 and LHX3 in SM neurons: ISL1 and LHX3 synergized to expand GFP activity to the dorsal spinal cord where additional MNR2 (ISL1 + LHX3, 19.5 ± 2.5 cells, p < 0.001) and ckSlit2 mRNAs were found, while ISL1 or LHX3 alone did not (arbitrary unit of Slit2 intensity; control, 1.0 ± 0.0 fold; ISL1, 0.9 ± 0.1 fold; LHX4, 0.9 ± 0.1 fold; ISL1 + LHX3, 1.2 ± 0.1 fold; ISL1 + LHX4, 1.3 ± 0.1 fold, p < 0.001) (Fig. 5R–U,W–Z,BB–EE,HH,JJ,LL). LHX4 had similar effect to LHX3 (ISL1 + LHX4, 26.6 ± 7.5 cells, p < 0.001), indicating that the two LHX factors may play redundant roles in SM neurons (Fig. 5V,AA,FF). We conclude that ISL1 and LHX3 are sufficient to generate SM neuronal traits and induce Slit2 transcription in the spinal cord.

ISL1-LHX3/4 complexes activate the Slit2 enhancer.
(A) ChIP-seq analysis of ISL1 and LHX3 binding to the Slit2 genome. Red arrow indicates Slit2 enhancer. (B–Q) GFP expression in transverse sections of HH stage 24 chick embryos electroporated with the Slit2 enhancer: GFP reporter, the CMV::mCherry vector as an internal control, ISL1, or LHX4 as indicated. Expression of Slit2, TBX20 and MNR2 was assessed in adjacent sections. In the hindbrain, the GFP signal is present in BM neurons (arrowheads), which have become medially expanded in the presence of ISL1 and LHX4 (brackets). Slit2 mRNA is induced in the same area (bracket), while the number of TBX20-expressing cells is unchanged. (R-FF) In the spinal cord, GFP activity is present in SM neurons. In the presence of ISL1-LHX3 or ISL1-LHX4, however, GFP expression and Slit2 mRNA have expanded dorsally together with ectopic MNR2-expressing cells (brackets) (>3 sections in 3 embryos in each group). (GG–LL) Quantification of GFP and Slit2 intensities, and TBX20 and MNR2-expressing cells (>3 sections in 3 embryos in each group). Error bars represent SEM. ***p < 0.001; Mann-Whitney rank sum test in GG (n = 4), unpaired Student’s t-test in (HH–LL) (n = 4); n.s., not significant. Scale bars: in (M), 100 μm for (B–M) in (Q), 100 μm for (N–Q) in (AA), 100 μm for (R-AA) in (FF), 100 μm for (BB–FF).
Since LIM-HD transcription factors bind to AT-rich sequences, we searched for binding sites of ISL1 and LHX3/4 in the Slit2 enhancer1,36,37. There were a few AT-rich motifs in the Slit2 enhancer and the enhancer was evolutionarily conserved in different species (Fig. 6A,B). When a luciferase reporter with the full length Slit2 enhancer 1–551 was transfected into HEK 293T cells, ISL1 and LHX3 (3.98 ± 0.20 fold, p < 0.001) and ISL1 and LHX4 (4.74 ± 0.37 fold, p < 0.001) induced strong transactivation of the reporter (Fig. 6C). When the activity of reporters with deletions was measured, only a reporter carrying region 1–270 (ISL1 + LHX3, 3.38 ± 0.26 fold; ISL1 + LHX4, 3.41 ± 0.17 fold, p < 0.001) but not ones carrying regions 1–184 or 271–551 was activated by ISL1-LHX4 or ISL1-LHX3, indicating that the ISL1-LHX3/4 binding sites lie within region 184–270. There are two AT-rich motifs within this region; we therefore mutated them individually to produce reporters mut1 and mut2 (Fig. 6C). Mut1 reporter activity was induced by ISL1-LHX3/4 (ISL1 + LHX3, 4.04 ± 0.46 fold; ISL1 + LHX4, 2.61 ± 0.39 fold, p < 0.001), but that of mut2 was not, indicating that ISL1-LHX3/4 binds to the region containing the mut2 site (Fig. 6C). We next introduced GFP reporters with deletions or point mutations into the spinal cord to test whether these reporters retained motor neuron-specific expression or not. Paralleling the results of the luciferase assays, we found that the reporters harboring regions 1–551 and 1–270, and the mut1 reporter produced normal motor neuron-specific GFP activity and were induced by ISL1-LHX3 (Fig. 6D,E,H,J,K,N). On the other hand, the reporters harboring regions 1–184 and 271–551 generated very little GFP and were not induced by ISL1-LHX3, confirming that they lacked the transcription factor binding sites essential for reporter activity (Fig. 6F,G,L,M). The mut2 reporter had also lost motor neuron-specific activity and was not induced by ISL1-LHX3 or ISL1-LHX4 (Fig. 6I,O,P). Together, these results show that the Slit2 enhancer is activated by ISL1 and LHX4 in BM neurons, and ISL1 and LHX3 in SM neurons (Fig. 6Q).

ISL1-LHX3 and ISL1-LHX4 binding sites in the Slit2 enhancer are evolutionarily conserved.
(A) Multiple sequence alignment of potential LIM-HD-binding sites (green and red texts) in the Slit2 enhancer in human, bonobo, mouse, chicken and frog. Point mutations introduced were shown below. (B) Phylogenetic tree of species in which Slit2 enhancers were analyzed. (C) Slit2 enhancer luciferase activity was measured in HEK 293T cells. Error bars represent s.e.m. ***p < 0.001, unpaired Student’s t-test (n > 3). (D–P) Activity of Slit2 GFP reporter derivatives measured by in ovo electroporation of chicks. MN-specific GFP activity was present in reporters with regions 1–551, 1–270 and mut1 but not in those with regions 1–184, 271–551 and mut2 (>11 sections in 3 embryos in each group). (Q) Model of the regulation of transcription regulation of the ISL1 and LHX factors during motor neuron development. The diagram of the ISL1-LHX hexamer complex is simplified. (R) Phylogenetic tree of species for which LHX3 and LHX4 protein sequences were analyzed. Scale bar: in (P), 100 μm for (D–P).
Discussion
Cranial motor neurons are divided into two subpopulations called BM/VM neurons and SM neurons with different origins and properties5. In this study, we demonstrated that BM/VM neurons did arise in Isl1 mutants but their BM neurons were defective to varying degrees in cell body migration and axon projection, supporting their combinatorial action.
ISL1 requires additional LIM-HD factor to form a multimeric complex for its function as demonstrated in the cortex (with LHX8) and spinal cord (with LHX3), which may serve different roles2,19,35. Among total 12 members, we speculated that LHX4 is the plausible LIM-HD transcription factor that works with ISL1 in BM neurons. LHX4 is present in both BM and SM neurons, while LHX3 is only present in SM neurons30,38,39,40,41,42,43,44,45. The presence of LHX4 in motor neurons has been known for a while but its role has been relatively underestimated due to its redundancy with LHX3 in SM neurons; only when both LHX3 and LHX4 are eliminated, SM neurons transfated to interneurons31,39,46,47. Interestingly, BM neurons such as spinal accessary motor column (SAC) cells remain in the absence of LHX3 and LHX4, which indicates that specification of BM neurons is intact without LHX439. We also observed that BM neurons were normally specified in the absence of ISL1, which together suggests that LHX4 is dispensable for specification of BM neurons. In this study, we demonstrated that both LHX3 and LHX4 have equivalent abilities to induce ectopic SM neurons when electroporated with ISL1 in the chick spinal cord. However, forced expression of ISL1 and LHX4 did not induce ectopic BM neurons or SM neurons in r4 chick hindbrains, indicating that potential of progenitors are already regionally-specified. Genome-wide analyses to distinguish BM and SM neuronal populations demonstrated that genome-wide binding sites of ISL1 differ in ESC-derived BM and SM neurons1,24. Moreover, ISL1 tends to bind adjacent to PHOX2 or LHX3 in BM and SM neurons, respectively1,24. Thus, different transcription profiles and environmental factors segregates BM and SM neurons, in which LHX4 behaves differently in cell fate and axon development2,8.
Several lines of evidence suggest that LIM codes control various functions in differentiating neurons, since the positioning of cell bodies, axon projections, neurotransmitter identities and ion channel expression, are all affected in many different neurons when ISL1 is downregulated9,23,48,49,50,51,52. Both ISL1 and LHX4 appeared to be required to induce Slit2 transcription as shown in cell lines, chick and mouse embryos, which favors the idea that they constitute the LIM code. Moreover we were able to locate their binding region in the intron of the Slit2 locus within the Slit2 enhancer. The activity of the Slit2 enhancer was highest only when both factors were present and mutating the putative binding sites abolished responsiveness. Furthermore, misexpression of ISL1 and LHX3/4 induced ectopic Slit2 transcription, raising the possibility that Slit2 is a direct target of ISL1 and LHX3/4. The molecular and genetic evidence that we have obtained indicates that different LIM codes assign different functions; BM neurons utilize the ISL1-LHX4 complex for axon development, while SM neurons employ ISL1-LHX3 and ISL1-LHX4 complexes to assign cell identity.
LHX4 is highly homologous to LHX3, with 66% identity at the amino acid level and 95% identity in the homeodomain, and they have similar bioactivity in vitro18,53,54,55. However, high resolution analysis of sequence preference revealed that, although the homeodomains of LHX3 and LHX4 have generally similar binding preferences, they have slightly different preferences for weaker motifs56. In addition, their differences in the LIM domain (about 82%), the interface that binds to other proteins, imply that subtle differences in this region may lead to divergence of their functions18. Previously we and others demonstrated that transcription is regulated differently in hindbrain and spinal motor neurons and is correlated with motor neuron diversification during evolution57,58. Thus, it is possible that LHX3 and LHX4 have evolved to share both common and distinct roles. They diverged recently during evolution: only one LHX3/4 factor is found in the lancelet and lamprey genome, in both of which hindbrain patterning is not yet fully established59 (Fig. 6R). The lancelet lacks a hindbrain, and in the lamprey the position of BM nuclei do not match with specific rhombomeres57,60,61. However, LHX3 and LHX4 have diverged in more advanced aquatic vertebrates in which hindbrain patterning is complete, i.e., BM nuclei align with rhombomere boundaries57. Thus, the segregation of LHX3 and LHX4 during evolution may suggest that different LHX factors serve different functions in SM and BM neurons.
In attempts to dissect out the genetic programs that define BM and SM neurons, several groups have compared the gene expression profiles of ESCs fated to become BM vs. SM neurons and found a large number that were either differentially enriched or similarly expressed in the two cell types1,24,35. For instance, a genome-wide Chip-seq analysis revealed that about 22–26% of genes were bound by ISL1 in both cell types while the rest were bound by ISL1 in only either of them1. However, when compared with microarray results from Isl1-deficient cells, only less than 50 genes were predicted to be targets of ISL1. When we examined mRNA expression of candidate genes by in situ hybridization, only transcript level of Slit2 transcripts was significantly reduced in BM neurons of Isl1 mutant mice. Nevertheless, more genes are likely to be controlled by Isl1, given the number of genes bound by Isl1 in ChIP-Seq analysis (1,590 genes) and the number of genes (683 genes) whose expression level was altered in our microarray results62. One possibility is that embryonic stem cells used in most studies may not fully represent cellular context of motor neurons in vivo, or in situ hybridization technique is not sensitive enough to detect subtle differences in gene expression level. More comprehensive transcriptome analysis in vivo will give us better clues to understand full repertoire of target genes controlled by ISL1 in developing motor neurons.
ROBO-SLIT signaling serves various developmental roles in the CNS including axon guidance, neuronal migration and axon and dendritic branching, mostly ‘within’ the neural tube. For instance, in the spinal cord, cell bodies and axons of motor neurons cross the midline when Slit or Robo genes are downregulated28,63. Similarly, in the hindbrain, errors in central projection and migration of BM neurons have been reported in Slit and Robo mutants: IEE neurons fail to cross the midline and trigeminal axons project in ectopic locations, and cell bodies of FBM neurons abnormally cross the midline25,28. However, the role of ROBO-SLIT signaling in peripheral projections is still less understood. It is reported that ROBO receptors control on peripheral projections of BM/VM neurons and their neurites respond to SLIT ligands in vitro25. This is reminiscent of ROBO-SLIT signaling in spinal cord motor axons; both SLIT2 and ROBO receptors are present in motor axons and transmit ROBO-SLIT signaling in an autocrine/juxtaparacrine manner64. It is still uncertain by which mechanisms Slit2 expression in them influence axons of BM/VM neurons. There are several locations in the developing CNS in which a ligand and its receptor co-exist within the same population65,66,67,68. For instance, altering intrinsic SEMA3A levels affected motor axon trajectories and the sensitivity of growth cones to exogenous semaphorins, probably because the endogenous ligand masked the receptor on the cell surface or influenced its trafficking69. Alternatively, the presence of a ligand may affect receptor activity, i.e., phosphorylation or downstream signaling, as shown in EPHRIN/EPH and SEMA6A/PLEXINA4 interactions66,68. It is not clear whether SLIT ligands control the availability of ROBO receptors or influence downstream pathways shared with other navigation cues. Nevertheless, our results demonstrate that ISL1 and LHX factors control motor neuron identity and Slit2-transcription in a cell-type-specific manner.
Methods
Mice
Isl1 hypo, Isl1 null, Isl1 flox mice, ISLMN: GFP-F, and Hb9::GFP mice were described previously8,10,14,35,63. Nestin-Cre mice were obtained from Jackson laboratory. Wildtype C56BL/6 mice (6–8 weeks old) were purchased from Damul Science. All experiments used protocols approved by the Animal Care and Ethics Committees of the Gwangju Institute of Science and Technology (GIST) in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The day when a vaginal plug was detected was designated embryonic day 0.5 (E0.5).
DNA constructs
Mouse Slit2 enhancer (chr5:48181677-48182228) was amplified by PCR using the genomic DNA from mouse. PCR fragments were subcloned into the pCS2 mini CMV-GFP and pCS2 mini CMV-nucGFP which contains a 60 bp TATA box and the transcription initiation site of the cytomegalovirus (CMV) promoter. The mini CMV promoter and EGFP sequences were amplified by PCR from pEGFP-N1 (Clontech). For luciferase assay, Slit2 enhancer PCR fragments were subcloned into the tk-luciferase reporter vector (Clontech). Isl1 and Lhx3 plasmids were described previously8. Mouse Lhx4 was amplified by PCR using mouse cDNA and subcloned into the pCAGGS1 vector. Fragments of mouse Slit2 enhancer 1–270, 1–184, 271–551 were amplified by PCR and mutations were introduced in mouse Slit2 enhancer by PCR-based mutagenesis.
In ovo electroporation
Slit2 enhancer::GFP, Slit2 enhancer::nucGFP, pCAGGS1-mIsl1, pCAGGS1-mLhx3, pCAGGS1-mLhx4 and pmCherry-C1 (Clontech) were electroporated into the chick hindbrains and spinal cords at HH stages 10 to 12, using a square wave electroporator (BTX) with 5 pulses of 18 V, 50 ms at 1 s intervals. Embryos were harvested at HH stages 23 to 24.
Luciferase assays
HEK 293T cells were seeded and incubated for 24 hours, and transiently transfected with reporters and transcription factors using Lipofectamine 2000 reagent (Invitrogen). CMV-β-galactosidase plasmid was co-transfected to normalize transfection efficiency. Cells were harvested about 40 hours after transfection, and cell extracts were assayed for luciferase assays and β-galactosidase assays. Data represent as means of triplicate value and all transfections were repeated independently at least three times.
Immunohistochemistry and In situ hybridization
Immunohistochemistry or in situ hybridization was performed as described previously8. The following antibodies were used: rabbit and guinea pig anti-HB970, rabbit anti-GFP (Invitrogen), mouse anti-GFP (Sigma), guinea pig anti-CHX102, rabbit anti-TBX2071, rabbit anti-PHOX2B72, rabbit anti-ISL1/210, rabbit anti-LHX1/210, guinea pig anti-LHX373, goat anti-LHX9 (Santa Cruz Biotechology). For flat-mount or whole mount immunostaining, flat-mounted hindbrains or embryos were fixed in 4% PFA, permeablized and then processed for immunostaining71. For in situ hybridization, embryonic mouse cDNA at E11.5 was used to generate riboprobes using an Advantage cDNA PCR kit (Clontech).
Quantification
To quantify ISL1 immunoreactivity in FBM neurons, 12 μm-thick transverse sections of r4 hindbrains were immunolabeled with anti-ISL1/2 and PHOX2B antibodies. The positions of Isl1-null FBM neurons were identified from the expression of PHOX2B. The background-subtracted pixel intensities of FBM nuclei were measured using MetaMorph software (Molecular Devices). To count the number of BM and SM neurons in each rhombomere, at least 3 sections from 3 embryos were analyzed for each group. The numbers of GFP, MNR2 and TBX20-expressing cells were determined in 12 μm-thick transverse sections after immunohistochemical staining. The number of nucGFP-expressing cells in the hindbrain was measured in the region medial to FBM nucleus in r2 and r4, as defined by TBX20 expression in the same sections. The level of ckSlit2 transcripts in the medial region of hindbrain and 250 × 650 pixel areas in the dorsal spinal cord were measured in adjacent transverse sections using ImageJ software. In the spinal cord, the background-subtracted pixel intensities of GFP in 250 × 650 pixel areas in the dorsal spinal cord were measured using ImageJ. At least 9 sections from 3 chick embryos were analyzed in each group. Statistical significance was analyzed by unpaired Student’s t-test or Mann-Whitney rank sum test as indicated in figure legends. To measure the length of trigeminal and FBM axons, peripheral projections from the exit point to the nerve terminals were manually traced. To calculate mean axon thickness, axon bundles were divided into 5 parts from proximal to distal and thickness at each intersection was averaged. All quantifications in images were analyzed in MetaMorph software (Molecular Devices).
Bioinformatic analysis
Enhancer sequences from human, bonobo, mouse, chicken and frog were retrieved from UCSC genome browser and aligned with mVISTA (genome.lbl.gov/vista) using the LAGAN alignment tool. LHX3/4 protein sequences from human, bonobo, mouse, chicken, frog, zebrafish, fugu, and lancelet were retrieved from NCBI protein database and lamprey sequence was retrieved from Japanese lamprey genome database. The LHX3/4 evolutionary history was inferred using the maximum parsimony method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown below the branches. The MP tree was obtained using the Subtree-Pruning-Regrafting (SPR) algorithm. The tree is drawn to scale, with branch lengths calculated using the average pathway method and are in the units of the number of changes over the whole sequences. Evolutionary analyses were conducted in MEGA6. For microarray analysis, we considered gene expression change as significant if the change of expression was ≥2 fold in genes down-regulated in Isl1-deficient ESC-derived motor neurons and ≥2.5 fold in genes induced in NesE-PHOX2B ESC-derived VM neurons and NesE-OLIG2 ESC-derived SM neurons23,24. To search for direct downstream targets of ISL1, we selected up to 1.5-fold binding peaks within ± 2 kb of gene boundaries in previously published dataset1.
Additional Information
How to cite this article: Kim, K.-T. et al. ISL1-based LIM complexes control Slit2 transcription in developing cranial motor neurons. Sci. Rep. 6, 36491; doi: 10.1038/srep36491 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Mazzoni, E. O. et al. Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat Neurosci 16, 1219–1227 (2013).
Thaler, J. P., Lee, S. K., Jurata, L. W., Gill, G. N. & Pfaff, S. L. LIM factor Lhx3 contributes to the specification of motor neuron and interneuron identity through cell-type-specific protein-protein interactions. Cell 110, 237–249 (2002).
Pattyn, A., Hirsch, M., Goridis, C. & Brunet, J. F. Control of hindbrain motor neuron differentiation by the homeobox gene Phox2b. Development 127, 1349–1358 (2000).
Varela-Echavarria, A., Tucker, A., Puschel, A. W. & Guthrie, S. Motor axon subpopulations respond differentially to the chemorepellents netrin-1 and semaphorin D. Neuron 18, 193–207 (1997).
Guthrie, S. Patterning and axon guidance of cranial motor neurons. Nat Rev Neurosci 8, 859–871 (2007).
Varela-Echavarria, A., Pfaff, S. L. & Guthrie, S. Differential expression of LIM homeobox genes among motor neuron subpopulations in the developing chick brain stem. Mol Cell Neurosci 8, 242–257 (1996).
Hutchinson, S. A. & Eisen, J. S. Islet1 and Islet2 have equivalent abilities to promote motoneuron formation and to specify motoneuron subtype identity. Development 133, 2137–2147 (2006).
Song, M. R. et al. Islet-to-LMO stoichiometries control the function of transcription complexes that specify motor neuron and V2a interneuron identity. Development 136, 2923–2932 (2009).
Tanaka, H. et al. Islet1 selectively promotes peripheral axon outgrowth in Rohon-Beard primary sensory neurons. Dev Dyn 240, 9–22 (2011).
Pfaff, S. L., Mendelsohn, M., Stewart, C. L., Edlund, T. & Jessell, T. M. Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell 84, 309–320 (1996).
Segawa, H. et al. Functional repression of Islet-2 by disruption of complex with Ldb impairs peripheral axonal outgrowth in embryonic zebrafish. Neuron 30, 423–436 (2001).
Thor, S. & Thomas, J. B. The Drosophila islet gene governs axon pathfinding and neurotransmitter identity. Neuron 18, 397–409 (1997).
Pan, L., Deng, M., Xie, X. & Gan, L. ISL1 and BRN3B co-regulate the differentiation of murine retinal ganglion cells. Development 135, 1981–1990 (2008).
Sun, Y. et al. A central role for Islet1 in sensory neuron development linking sensory and spinal gene regulatory programs. Nat Neurosci 11, 1283–1293 (2008).
Agulnick, A. D. et al. Interactions of the LIM-domain-binding factor Ldb1 with LIM homeodomain proteins. Nature 384, 270–272 (1996).
Bach, I., Carriere, C., Ostendorff, H. P., Andersen, B. & Rosenfeld, M. G. A family of LIM domain-associated cofactors confer transcriptional synergism between LIM and Otx homeodomain proteins. Genes & development 11, 1370–1380 (1997).
Jurata, L. W., Pfaff, S. L. & Gill, G. N. The nuclear LIM domain interactor NLI mediates homo- and heterodimerization of LIM domain transcription factors. J Biol Chem 273, 3152–3157 (1998).
Gadd, M. S. et al. Structural basis for partial redundancy in a class of transcription factors, the LIM homeodomain proteins, in neural cell type specification. J Biol Chem 286, 42971–42980 (2011).
Cho, H. H. et al. Isl1 directly controls a cholinergic neuronal identity in the developing forebrain and spinal cord by forming cell type-specific complexes. PLoS Genet 10, e1004280 (2014).
Lewcock, J. W., Genoud, N., Lettieri, K. & Pfaff, S. L. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56, 604–620 (2007).
Kraus, F., Haenig, B. & Kispert, A. Cloning and expression analysis of the mouse T-box gene tbx20. Mech Dev 100, 87–91 (2001).
Karis, A. et al. Transcription factor GATA-3 alters pathway selection of olivocochlear neurons and affects morphogenesis of the ear. The Journal of comparative neurology 429, 615–630 (2001).
Lee, H. et al. Slit and Semaphorin signaling governed by Islet transcription factors positions motor neuron somata within the neural tube. Exp Neurol 269, 17–27 (2015).
Panman, L. et al. Transcription factor-induced lineage selection of stem-cell-derived neural progenitor cells. Cell Stem Cell 8, 663–675 (2011).
Hammond, R. et al. Slit-mediated repulsion is a key regulator of motor axon pathfinding in the hindbrain. Development 132, 4483–4495 (2005).
Kidd, T., Bland, K. S. & Goodman, C. S. Slit is the midline repellent for the robo receptor in Drosophila. Cell 96, 785–794 (1999).
Ma, L. & Tessier-Lavigne, M. Dual branch-promoting and branch-repelling actions of Slit/Robo signaling on peripheral and central branches of developing sensory axons. J Neurosci 27, 6843–6851 (2007).
Kim, M. et al. Motor neuron cell bodies are actively positioned by Slit/Robo repulsion and Netrin/DCC attraction. Dev Biol 399, 68–79 (2015).
Chen, Z., Gore, B. B., Long, H., Ma, L. & Tessier-Lavigne, M. Alternative splicing of the Robo3 axon guidance receptor governs the midline switch from attraction to repulsion. Neuron 58, 325–332 (2008).
Gavalas, A., Ruhrberg, C., Livet, J., Henderson, C. E. & Krumlauf, R. Neuronal defects in the hindbrain of Hoxa1, Hoxb1 and Hoxb2 mutants reflect regulatory interactions among these Hox genes. Development 130, 5663–5679 (2003).
Li, H. et al. Gsh-4 encodes a LIM-type homeodomain, is expressed in the developing central nervous system and is required for early postnatal survival. EMBO J 13, 2876–2885 (1994).
Amin, N. D. et al. Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 350, 1525–1529 (2015).
Lee, S. et al. STAT3 promotes motor neuron differentiation by collaborating with motor neuron-specific LIM complex. Proc Natl Acad Sci USA 110, 11445–11450 (2013).
Thiebes, K. P. et al. miR-218 is essential to establish motor neuron fate as a downstream effector of Isl1-Lhx3. Nat Commun 6, 7718 (2015).
Lee, S. K., Jurata, L. W., Funahashi, J., Ruiz, E. C. & Pfaff, S. L. Analysis of embryonic motoneuron gene regulation: derepression of general activators function in concert with enhancer factors. Development 131, 3295–3306 (2004).
Lee, S. et al. Fusion protein Isl1-Lhx3 specifies motor neuron fate by inducing motor neuron genes and concomitantly suppressing the interneuron programs. Proc Natl Acad Sci USA 109, 3383–3388 (2012).
Machinis, K. & Amselem, S. Functional relationship between LHX4 and POU1F1 in light of the LHX4 mutation identified in patients with pituitary defects. J Clin Endocrinol Metab 90, 5456–5462 (2005).
Ando, H. et al. Lhx2 mediates the activity of Six3 in zebrafish forebrain growth. Dev Biol 287, 456–468 (2005).
Sharma, K. et al. LIM homeodomain factors Lhx3 and Lhx4 assign subtype identities for motor neurons. Cell 95, 817–828 (1998).
Grillet, N., Dubreuil, V., Dufour, H. D. & Brunet, J. F. Dynamic expression of RGS4 in the developing nervous system and regulation by the neural type-specific transcription factor Phox2b. J Neurosci 23, 10613–10621 (2003).
Bami, M., Episkopou, V., Gavalas, A. & Gouti, M. Directed neural differentiation of mouse embryonic stem cells is a sensitive system for the identification of novel Hox gene effectors. PLoS One 6, e20197 (2011).
Jackman, W. R., Draper, B. W. & Stock, D. W. Fgf signaling is required for zebrafish tooth development. Dev Biol 274, 139–157 (2004).
Muller, M., Jabs, N., Lorke, D. E., Fritzsch, B. & Sander, M. Nkx6.1 controls migration and axon pathfinding of cranial branchio-motoneurons. Development 130, 5815–5826 (2003).
Chizhikov, V. V. et al. Lmx1a regulates fates and location of cells originating from the cerebellar rhombic lip and telencephalic cortical hem. Proc Natl Acad Sci USA 107, 10725–10730 (2010).
Ding, Y. Q. et al. Lmx1b is essential for the development of serotonergic neurons. Nat Neurosci 6, 933–938 (2003).
Ericson, J. et al. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell 90, 169–180 (1997).
Seredick, S., Hutchinson, S. A., Van Ryswyk, L., Talbot, J. C. & Eisen, J. S. Lhx3 and Lhx4 suppress Kolmer-Agduhr interneuron characteristics within zebrafish axial motoneurons. Development 141, 3900–3909 (2014).
Bejarano-Escobar, R. et al. Expression and function of the LIM-homeodomain transcription factor Islet-1 in the developing and mature vertebrate retina. Exp Eye Res 138, 22–31 (2015).
Liang, X. et al. Isl1 is required for multiple aspects of motor neuron development. Mol Cell Neurosci 47, 215–222 (2011).
Wolfram, V. et al. The transcription factors islet and Lim3 combinatorially regulate ion channel gene expression. J Neurosci 34, 2538–2543 (2014).
Kohl, A., Marquardt, T., Klar, A. & Sela-Donenfeld, D. Control of axon guidance and neurotransmitter phenotype of dB1 hindbrain interneurons by Lim-HD code. J Neurosci 35, 2596–2611 (2015).
Shirasaki, R., Lewcock, J. W., Lettieri, K. & Pfaff, S. L. FGF as a target-derived chemoattractant for developing motor axons genetically programmed by the LIM code. Neuron 50, 841–853 (2006).
Sloop, K. W., Dwyer, C. J. & Rhodes, S. J. An isoform-specific inhibitory domain regulates the LHX3 LIM homeodomain factor holoprotein and the production of a functional alternate translation form. J Biol Chem 276, 36311–36319 (2001).
Kawamata, N. et al. A novel chromosomal translocation t (1;14) (q25; q32) in pre-B acute lymphoblastic leukemia involves the LIM homeodomain protein gene, Lhx4. Oncogene 21, 4983–4991 (2002).
West, B. E. et al. Regulation of the follicle-stimulating hormone beta gene by the LHX3 LIM-homeodomain transcription factor. Endocrinology 145, 4866–4879 (2004).
Berger, M. F. et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133, 1266–1276 (2008).
Murakami, Y. et al. Segmental development of reticulospinal and branchiomotor neurons in lamprey: insights into the evolution of the vertebrate hindbrain. Development 131, 983–995 (2004).
Kim, N., Park, C., Jeong, Y. & Song, M. R. Functional Diversification of Motor Neuron-specific Isl1 Enhancers during Evolution. PLoS Genet 11, e1005560 (2015).
Hunter, C. S. & Rhodes, S. J. LIM-homeodomain genes in mammalian development and human disease. Mol Biol Rep 32, 67–77 (2005).
Holland, P. W. & Garcia-Fernandez, J. Hox genes and chordate evolution. Dev Biol 173, 382–395 (1996).
Holland, L. Z. & Holland, N. D. Chordate origins of the vertebrate central nervous system. Curr Opin Neurobiol 9, 596–602 (1999).
Lee, H. et al. Slit and semaphorin signaling governed by Islet transcription factors positions motor neuron somata within the neural tube. Exp. Neurol. 269, 17–27 (2015).
Bai, G. et al. Presenilin-dependent receptor processing is required for axon guidance. Cell 144, 106–118 (2011).
Jaworski, A. & Tessier-Lavigne, M. Autocrine/juxtaparacrine regulation of axon fasciculation by Slit-Robo signaling. Nat Neurosci 15, 367–369 (2012).
Triplett, J. W. & Feldheim, D. A. Eph and ephrin signaling in the formation of topographic maps. Semin Cell Dev Biol 23, 7–15 (2012).
Carvalho, R. F. et al. Silencing of EphA3 through a cis interaction with ephrinA5. Nat Neurosci 9, 322–330 (2006).
Hornberger, M. R. et al. Modulation of EphA receptor function by coexpressed ephrinA ligands on retinal ganglion cell axons. Neuron 22, 731–742 (1999).
Haklai-Topper, L., Mlechkovich, G., Savariego, D., Gokhman, I. & Yaron, A. Cis interaction between Semaphorin6A and Plexin-A4 modulates the repulsive response to Sema6A. EMBO J 29, 2635–2645 (2010).
Moret, F., Renaudot, C., Bozon, M. & Castellani, V. Semaphorin and neuropilin co-expression in motoneurons sets axon sensitivity to environmental semaphorin sources during motor axon pathfinding. Development 134, 4491–4501 (2007).
Thaler, J. et al. Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron 23, 675–687 (1999).
Song, M. R. et al. T-Box transcription factor Tbx20 regulates a genetic program for cranial motor neuron cell body migration. Development 133, 4945–4955 (2006).
Pattyn, A., Morin, X., Cremer, H., Goridis, C. & Brunet, J. F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 399, 366–370 (1999).
Thaler, J. P. et al. A postmitotic role for Isl-class LIM homeodomain proteins in the assignment of visceral spinal motor neuron identity. Neuron 41, 337–350 (2004).
Acknowledgements
We thank Dr. Sally A. Camper at the University of Michigan for Lhx4 knock-out samples, and Drs. Woo Jin Park and Minkyung Kim for technical help and comments. Support for M-RS and this research was provided by grants from NRF (2013R1A1A2058548), KHIDI HI14C3484 and the GIST Research Institute (GRI) in 2016. This project was supported by NIH RO1 NS054740 and R21 NS077169 to GSM. Use of the Nevada Genomics Center was supported by P20 RR-016464 from INBRE (NCRR). Use of University of Nevada tissue culture and imaging core facilities was supported by P20 RR-016464, P20 GM103440, P20 GM103554, and P20 GM103650.
Author information
Authors and Affiliations
Contributions
K.-T.K. and M.-R.S. conceived the project, designed the experiments and drafted the manuscript. K.-T.K. carried out the experiments, with the exception of analysis of Robo1/2 mutant mice and bioinformatics analysis. N.K., H.-K.K., H.L. and H.-C.P. contributed the analysis of animal models and technical support. H.N.G and G.S.M. contributed to the analysis of the Robo1/2 mutant mice. P.G. provided Lhx4 mutant mice samples and technical support. C.P. performed bioinformatic analysis.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kim, KT., Kim, N., Kim, HK. et al. ISL1-based LIM complexes control Slit2 transcription in developing cranial motor neurons. Sci Rep 6, 36491 (2016). https://doi.org/10.1038/srep36491
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36491
This article is cited by
-
Multi-omic analysis of selectively vulnerable motor neuron subtypes implicates altered lipid metabolism in ALS
Nature Neuroscience (2021)
-
Impairment of motor coordination and interneuron migration in perinatal exposure to glufosinate-ammonium
Scientific Reports (2020)
-
LIM homeodomain transcription factor Isl1 affects urethral epithelium differentiation and apoptosis via Shh
Cell Death & Disease (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
