
Abstract
NLRP3 (NOD-like receptor family, pyrin domain containing 3) is a member of the inflammasome family and is of special interest in renal disease. Experimental studies have shown that Nlrp3 plays a significant role in the induction of renal damage and dysfunction in acute and chronic renal injury. However, the role of NLRP3 in human renal disease is completely unknown. From a retrospective cohort study, we determined in 1271 matching donor and recipient samples if several NLRP3 single nucelotide polymorphisms (SNPs) were associated with primary non-function (PNF), delayed graft function (DGF), biopsy-proven acute rejection (BPAR) and death-censored graft and patient survival. NLRP3 gain-of-function SNP (rs35829419) in donors was associated with an increased risk of BPAR while NLRP3 loss-of-function SNP (rs6672995) in the recipient was associated with a decreased risk of BPAR in the first year following renal transplantation (HR 1.91, 95% CI 1.38–2.64, P < 0.001 and HR 0.73, 95% CI 0.55–0.97, P = 0.03 resp.). NLRP3 SNPs in both donor and recipient were not associated with PNF, DGF, graft survival or patient survival. We conclude that genetic variants in the NLRP3 gene affect the risk of acute rejection following kidney transplantation.
Similar content being viewed by others

Introduction
Improving long-term renal allograft survival remains a major challenge and allograft longevity is affected by factors including donor type and age, kidney preservation method, ischemia-reperfusion injury (IRI) and occurrence of acute rejection1. Because the pool of living (un)related donor kidneys (LD) is limited, the majority of renal allografts are recovered from deceased brain dead and deceased cardiac dead donors. Unfortunately, the latter two groups display more IRI and inferior transplant outcome compared to LD2. IRI leads to necrosis, local and systemic immune activation and it contributes to delayed graft function (DGF), which in turn is associated with higher rates of acute rejection and affect graft survival. Innate immunity plays an important role in the mechanisms underlying IRI. Inflammasomes are innate immune sensors and monitor for signs of infection and tissue damage3,4. Protein-complex formation of inflammasome member NLRP3 (NOD-like receptor family, pyrin domain containing 3) with ASC (Apoptosis-associated Speck-like protein containing a Caspase activation and recruitment domain) and Caspase-1 leads to formation of active IL-1β, IL-18 and IL-33 and, through non-canonical inflammasome signalling, to a programmed form of cell death called pyroptosis3,4,5,6,7. NLRP3 is the best-characterized inflammasome and is of special interest in renal diseases. NLRP3 is able to induce an inflammatory response upon activation by endogenous stress ligands called damage-associated molecular patterns (DAMPs), which are released following tissue injury, including IRI. In addition, NLRP3 is highly expressed in murine and human renal epithelial cells and leukocytes and increasing amounts of experimental data show that Nlrp3 deficiency prevents renal dysfunction, damage and inflammation in several murine acute and chronic renal disease models8,9,10,11,12. Genotyping of SNPs (single nucleotide polymorphisms) is frequently used to determine the effects of genetic variations in the context of human diseases. Determination of NLRP3 SNPs in donor and recipient DNA offers an unique opportunity to characterize the effects of transplant-associated NLRP3 compared to the host’s infiltrating leukocyte-associated NLRP3, respectively. So far it is known that NLRP3 SNPs with a gain-of-function (GOF) are related to several auto-inflammatory disorders due to an enhanced inflammatory state in these patients13,14,15,16. Currently, it is unknown if NLRP3 SNPs are related to human renal diseases or patient outcome after solid organ transplantation5. Therefore, we determined NLRP3 SNPs in a large cohort of matching donors and recipients and investigated their association with renal allograft and patient outcomes.
Results
NLRP3 SNP distribution in donors and recipients
Baseline characteristics of donors and recipients are displayed in Table 1. In our cohort, genotypic distribution of NLRP3-related SNPs in donor and recipient were comparable to previous general/Caucasian population distributions (Supplementary Table S1 and www.ensemble.org). None of the SNPs deviated significantly from a Hardy-Weinberg equilibrium (all P > 0.46 after Holm-Bonferroni correction). In the combined model (A/A vs. A/a + a/a) genotype distribution of NLRP3 SNPs were comparable between donors and recipient and SNPs were not considered as a potential risk factor for diseases of the native kidneys. SNPs with minor allele frequency less than 1% were left out from further analysis (N = 3).
NLRP3 SNPs are not associated with delayed graft function, primary non-function, graft survival or patient survival
Delayed graft function (DGF) occurred in 415/1271 patients (33%) of which 60/415 resulted in primary non-functioning (PNF) of the graft (14% of DGF, 5% of total patients). None of the 3 NLRP3 SNPs in either donor or recipient were correlated to the occurrence of DGF or PNF (Table 2). After stratification by donor type (living and cadaveric), again none of the NLRP3 SNPs were significantly associated with DGF or PNF (Supplementary Table S2). Median overall graft survival was 5.5 years (interquartile range 2.9–8.9 years). The overall cumulative incidence of death-censored graft failure was 215/1271 (17%) of which 136/215 due to rejection (63% of graft failure, 11% of total). We did not observe a significant association between any of the NLRP3 SNPs, in either donor or recipient, with death-censored graft failure (Table 2). The cumulative incidence of patient mortality was 220/1271 (17%). Donor and recipient NLRP3 SNPs were not associated with patient survival (Table 2).
Genetic variants in NLRP3 are a risk factor for biopsy-proven acute rejection
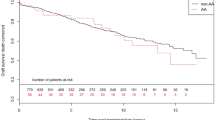
The overall cumulative incidence of biopsy-proven acute rejection (BPAR) was 34% (433/1271). The NLRP3 SNP rs35829419 (GOF) in the donor (A/a + a/a) was significantly associated with an increased risk for BPAR in the complete follow-up (HR 1.48, 95% CI 1.12–1.96, P = 0.006; Table 3 and Fig. 1), which was observed in cadaveric, but not in living donors after stratification for donor type (cadaveric donor HR 1.58, 95% CI 1.16–2.16, P = 0.004 vs. living donor HR 1.16, 95% CI 0.62–2.18, P = 0.64). NLRP3 SNP rs6672995 (LOF) in the recipient (A/a + a/a) was significantly associated with a decreased risk for BPAR in the complete follow-up (HR 0.72, 95% CI 0.58–0.91, P = 0.005; Table 3 and Fig. 1) which was observed in cadaveric, but not living donor after stratification for donor type (cadaveric donor HR 0.69, 95% CI 0.53–0.90, P = 0.005 vs. living donor HR 0.83, 95% CI 0.53–1.30, P = 0.42). From the Kaplan-Meier curves we could observe that BPAR occurred particularly within the first year after transplantation (Fig. 1). Therefore, we also stratified the patients in those who experienced a BPAR within the first 12 months after transplantation (early BPAR, N = 393 events) and those who experienced their first episode of BPAR later than 12 months after transplantation (late BPAR, N = 40 events). NLRP3 SNP rs35829419 in the donor was significantly associated with early, but not late BPAR (early BPAR HR 1.54, 95% CI 1.15–2.05, P = 0.003 vs. late BPAR HR 0.82, 95% CI 0.25–2.67, P = 0.74; Table 3). Similarly, NLRP3 SNP rs6672995 in the recipient was associated with early, but not late BPAR (early BPAR HR 0.68, 95% CI 0.54–0.87, P = 0.002 vs. late BPAR HR 1.16, 95% CI 0.60–2.25, P = 0.66; Table 3). Little differences in donor type distribution were observed for rs6672995 SNP allele combinations in recipients (Table S3).

Kaplan-Meier curves for biopsy-proven acute rejection-free survival in donor and recipient NLRP3 single nucleotide polymorphisms.
Kaplan-Meier curves for rejection-free survival of (A) donor NLRP3 single nucleotide polymorphism (SNP) rs35829419 and (B) recipient NLRP3 SNP rs6672995. The homozygous dominant genotypes (A/A) are depicted in blue and the combination of the heterozygous (A/a) and homozygous recessive (a/a) genotypes are shown in green. The P-value is calculated by log-rank testing.
Donor rs35829419 and recipient rs6672995 NLRP3 variants are independently associated with biopsy-proven acute rejection
Next, we created multivariate models adjusting for parameters that are known risk factors for BPAR from the literature, including donor and recipient age, recipient gender, type of donation (all without missing values), cold ischemia time (0.4% missing values), number of HLA mismatches (0.6% missing), previous transplantation (0.1% missing), pre-transplant panel reactive antibodies (3.0% missing), pre-transplant dialysis time (3.3% missing) and the use of induction therapy with interleukin-2-receptor antagonists (no missing values). In separate multivariate regression models, donor NLRP3 SNP rs35829419 (HR 1.37, 95% CI 1.01–1.87, P = 0.04; Table 4) and recipient NLRP3 SNP rs6672995 (HR 0.71, 95% CI 0.55–0.92, P = 0.01; Table 4) were independently associated with BPAR on complete follow-up. When we created a multivariate regression model that included both the donor rs35829419 and recipient rs6672995 NLRP3 SNPs in one analysis, both variants were contributors to the occurrence of BPAR (HR 1.34, 95% CI 0.99–1.83, P = 0.06 and HR 0.71, 95% CI 0.55–0.93, P = 0.01, respectively; light-gray area Table 4). When we performed mutliple imputations and recalculated the multivariable model including both SNPs, similar results were obtained (HR 1.38, 95% CI 1.04–1.83, P = 0.02 and HR 0.75, 95% CI 0.60–0.94, P = 0.01, respectively). By 10x internal cross-validation, these results were calculated to be 9.9% over-optimistic. Comparable results were obtained when we only investigated the cumulative incidence of BPAR within the first 12 months after transplantation (donor rs35829419 HR 1.42, 95% CI 1.04–1.94, P = 0.03 and recipient rs6672995 HR 0.67, 95% CI 0.51–0.89, P = 0.005; light-gray area Table 4). Again, when we performed mutliple imputations and recalculated the multivariable model including both SNPs, similar results were obtained (HR 1.41, 95% CI 1.06–1.89, P = 0.02 and HR 0.71, 95% CI 0.56–0.91, P = 0.006, respectively). By 10x internal cross-validation, these results were calculated to be 10.5% over-optimistic. In conclusion, these results indicate that NLRP3 SNPs rs35829419 (GOF) in the donor and rs6672995 (LOF) in the recipient both contribute to the risk of BPAR, and more specifically to those episodes of BPAR that occur within the first year after transplantation.
Discussion
The NLRP3 inflammasone is of special interest in renal diseases because of its expression pattern in murine and human kidneys and its detrimental role in experimental models of acute and chronic renal injury8,9,10,11,12. Thus far, nothing is known about the role of NLRP3 in human renal diseases or solid organ transplantation. Determination of NLRP3 SNPs in donor and recipient DNA offers a unique opportunity to characterize the differential effects of transplant- and the host’s leukocyte-associated NLRP3 inflammasome. We quantified several NLRP3 SNPs in a large cohort consisting of >1200 matched donors and recipients. We found that the NLRP3 gain-of-function SNP rs35829419 in donors associated with an increased risk of acute rejection, whereas on the contrary that the NLRP3 loss-of-function SNP rs6672995 in recipients associated with a reduced risk of rejection, particularly those episodes that occurred within the first year after transplantation.
NLRP3 SNPs became of particular interest in human disease because of their association and causal role in several autoinflammatory disorders. Gain-of-function mutations in the NLRP3 gene (R260W and Q705K) lead to enhanced basal IL-1β production and are linked to Muckle-Well syndrome, familial cold autoinflammatory syndrome and chronic infantile neurologic cutaneous and articular syndrome13,14,15,16. Moreover, NLRP3 SNP Q705K is associated with Crohn’s disease, celiac disease and late-onset Alzheimer disease17,18,19. In contrast, OR2B11/NLRP3 SNP rs4353135 and NLRP3 SNP rs6672995 lead to a reduction in basal NLRP3 expression and rs6672995 homozygous variant a/a affects IL-1β production. Both SNPs are therefore considered to cause a loss of the inflammasome’s function20. So far, knowledge about the role of NLRP3 in renal diseases is largely acquired from murine studies, where an important role for both tissue- and myeloid-related Nlrp3 in murine IRI was observed8,9,10. The literature suggests that the pathophysiological role of Nlrp3 in murine renal IRI is in part inflammasome-independent8,9,21,22,23 and might also be related to TGF-β signaling24.
We observed an increased risk of rejection with the donor gain-of-function NLRP3 variant Q705K (rs35829419) and a reduced risk of rejection in recipients carrying the NLRP3 A71T variant (rs6672995), especially within the first year following transplantation. It is known that episodes of acute rejection that occur early after transplantation are less detrimental to the graft than those that occur at later time-points. Chronic rejection, especially the antibody-mediated variant, is more challenging to treat. Since we observed an association of NLRP3 SNPs with early, but not late BPAR, this could explain why we did not identify an association with graft loss.
Little is known about the role of NLRP3 in adaptive immunity. Activation of the NLRP3 inflammasome is crucial in IL-1β and IL-18 maturation that in turn, besides their role in innate immunity, play an important part in priming of adaptive (allo)immunity. Both cytokines are involved in development and differentiation of CD4+ T-helper lymphocytes (reviewed in refs 4 and 25). Nlrp3 is known to boost adaptive immunity in an inflammasome-dependent manner in respiratory failure26, but Nlrp3 deficiency does not affect alum-mediated adjuvant activity in mice challenged with human serum albumin vaccination27. To date, it is unknown if NLRP3 is differentially involved in cellular compared to humoral immunity in humans. How gain- and loss-of-function variants in the NLRP3 gene affect acute rejection remains speculative and is an area of future study. A limitation of our study is the lack of a standardized assays for the determination of donor-specific antibodies over the years and we therefore could not investigate whether the association between the NLRP3 variants differed for T cell- or antibody-mediated rejection. We were also unable to directly measure the effect of the NLRP3 variants on inflammasome-related cytokine production. The current study is the first to describe the association between the NLRP3 variants and acute rejection and we therefore acknowledge that other cohort studies should validate the generalizability and transportability of our findings.
Interestingly, the association of the NLRP3 gain-of-function specifically in the donor and NLRP3 loss-of-function specifically in the recipient with the development of rejection suggests that NLRP3 could be a double-edged sword in renal transplantation. NLPR3 is known to be expressed in human myeloid and lymphoid cells, but also in cells of the renal parenchyma, including tubular epithelial cells9,11,28,29. Tubular epithelial cells express NLRP3, IL-1β and IL-18 and a gain-of-function of NLRP3 could result in elevated levels of these cytokines leading to (local) chronic inflammation and chronic kidney injury11,13,14,15,16,30. Additionally, tubular epithelial NLPR3 could also contribute to kidney fibrosis directly. In experimental murine studies for instance, tubular epithelial Nlrp3 was involved in epithelial-mesenchymal transmission (EMT) via TGF-β signaling, which is believed to precede the development of interstitial fibrosis and tubular atrophy11. However, besides parenchymal cells, one should keep in mind that also immune cells like resident dendritic cells of donor origin are being transplanted and subsequently present alloimmune epitopes in draining lymph nodes and a gain-of-function of NLRP3 in these cells could enhance direct allorecognition, resulting in early rejection. NLRP3 loss-of-function in recipients on the other hand could alter IL-1β, IL-18 and IL-33 production by (infiltrating) leukocytes upon activation, which was shown to be the case for IL-1β20. This could lead to a dampened alloimmune response after transplantation. Further experimental studies are required to determine how NLRP3 is involved in alloimmune recognition.
Given the important role of Nlrp3 in murine models of IRI, one would expect a role for the human NLRP3 gene in delayed graft function. However, neither donor nor recipient NLRP3 SNPs associated with delayed graft function or primary nonfunction. This lack of association could be explained by several aspects. Although murine ischemia-reperfusion injury is also characterized by tubular necrosis, there are some distinct differences with delayed graft function, including differences in circulating leukocyte composition, a slightly different renal anatomy, hemodynamic response and the lack of immunosuppressive drugs in the murine IRI model, making a direct extrapolation of murine data to the human clinical setting not always possible31. In addition, delayed graft function is a clinical term describing the need for additional renal replacement therapy, which could have other causes besides IRI-related acute kidney injury rejection, drug toxicity and hyperkalemia32 making it a heterogeneous surrogate end-point.
In our study, we grouped A/a and a/a genotype together based on low numbers of the latter group but we do not know if A/a is phenotypically different from a/a and how it would affect our study. In donors, we did identify patients with a homozygous recessive rs35829419 variant. For this variant, Verma et al. showed that in vitro, the rs35829419 mutant monocytes had a significantly higher level of IL1-beta protein at baseline or after stimulation with the pro-pyroptotic substance alum (adjuvant)16. Additionally, Roberts et al. observed that in the homozygous recessive allele combination, there was an increased plasma concentration of IL1-beta33. It is difficult to extrapolate these results to our the findings in our cohort: (1) there was no heterozygous testing in the in vitro experiments by Verma et al., (2) they observed that the excretion of IL1-beta was only partly rescued by a pan-caspase inhibitor, indicating potential non-canonical, pyroptotic signalling in their experiments, (3) both studies measured the full IL1-beta and IL18 protein and not the active forms that are cleaved by the inflammasome complex. The loss of function NLRP3 variant rs6672995 was shown to correlate with the NLRP3 expression on the mRNA level34. They did not find an association with plasma concentrations of IL1-beta in patients with this variant. In line with this report, Villani et al. found that patients who were homozygous for the rs6672995 variant had a lower production of IL1-beta as compared to heterozygous patients20. Again no active IL1-beta (or other inflammasome related cytokines), pyroptosis and in this case no patients with a homozygous dominant variant were analyzed and we can therefore not extrapolate these results to our cohort.
In summary, we showed that various gain- and loss-of-function genetic variants in the NLRP3 gene differentially associate with biopsy-proven acute rejection, specifically within the first year after renal transplantation. These findings imply a complex role for NLRP3 in early allorecognition and rejection, which makes timing of interventions that aim at promoting or reducing NLRP3 inflammasome activity crucial.
Material and Methods
Patient genotyping
Samples were collected from a retrospective cohort as described before35,36. Between March 1993 and February 2008, 1271 matching donor and recipient peripheral blood mononuclear cells (PBMCs) were obtained from all available patients who underwent kidney transplantation at the University Medical Center Groningen, The Netherlands. The exclusion criteria were: cases of re-transplantation, combined kidney/pancreas or kidney/liver transplantation, technical problems during surgery, the unavailability of DNA and loss of the patients on follow-up (Fig. 2). The institutional ethical review board of the University Medical Center Groningen approved the study (METc 2014/077). Written informed consent was obtained from all patients. None of the living transplant donors were from a vulnerable population and all living donors provided written informed consent. In case of deceased donation, the donors provided informed consent when they registered their donation status and by law, no additional consent was needed. The study was conducted according to the principles of the declaration of Helsinki. Non-synonymous NLRP3 SNPs were chosen from NCBI (2010). In addition, we chose the NLRP3/OR2B11 rs4353135 SNP, located downstream of NLRP3 and upstream of olfactory receptor gene OR2B11, which has previously been shown to influence NLRP3 expression levels20. Genotyping of the selected SNPs (Supplementary Table 1) was performed using the Illumina VeraCode GoldenGate Assay kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Genotype clustering and calling were performed using Beadstudio Software (Illumina). The selected SNPs were not in linkage disequilibrium (r2 < 0.10; SNP Annotation and Proxy Search, Broad institute).

Flowchart of the included patients.
DNA isolation and quality control
Peripheral blood mononuclear cells were used to acquire donor and recipient DNA. DNA samples were validated for DNA concentrations using absorbance at 260 nm with a spectrophotometer (ND-1000, NanoDrop) and purity was assessed by 260/280 and 260/230 absorbance ratios. In case of impurity of the sample, repeated isolation attempts were conducted.
Study end-points
The end-points used in this study were: delayed graft function (DGF, defined as the requirement for dialysis within the first week after transplantation), primary non function (PNF, defined as nonfunctioning of the allograft from transplantation on), time to the first episode of clinical biopsy-proven acute rejection (BPAR), death-censored graft failure (defined as the need for dialysis or re-transplantation) and patient mortality.
Statistics
Statistical analyses were performed using R version 2.15.2 for Macintosh (www.r-project.org). Two-sided P-values below 0.05 were considered statistically significant. As a routine data quality control, the Hardy-Weinberg equilibria for the SNPs were tested. Univariate P-values were subjected to Holm-Bonferroni correction for multiple testing. For the association with DGF and PNF, logistic regression models were constructed. For the association with BPAR, (rejection-mediated) death-censored graft failure and patient survival, time-to-event models were created by Cox proportional hazards regression. Data were considered missing (completely) at random and we therefore performed complete-case multivariable regression on 1014/1271 (79.8%) of patients with complete multivariate data available. To validate the effect of missing values on our multivariate models, we reperformed the Cox regression analyses on M = 20 imputed datasets by multiple imputations by chained equations (MICE). The imputation results from predictive mean matching were visually checked by Kernel density plots. Multivariate Cox models were subjected to 10x internal cross-validation and the decline in slope in the validation cohort was considered as an overestimation measure. Kaplan-Meier survival curves were generated to visualize the association between the genotypes of each SNP in donor and recipient and the time to BPAR. Due to the low numbers of patients with homozygous recessive (a/a) variants, we used co-dominant models in which we compared homozygous dominant (A/A) versus heterozygous + homozygous recessive (A/a + a/a) individuals.
Additional Information
How to cite this article: Dessing, M. C. et al. Donor and recipient genetic variants in NLRP3 associate with early acute rejection following kidney transplantation. Sci. Rep. 6, 36315; doi: 10.1038/srep36315 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Pascual, M., Theruvath, T., Kawai, T., Tolkoff-Rubin, N. & Cosimi, A. B. Strategies to improve long-term outcomes after renal transplantation. N Engl J Med 346, 580–590 (2002).
Terasaki, P. I., Cecka, J. M., Gjertson, D. W. & Takemoto, S. High survival rates of kidney transplants from spousal and living unrelated donors. N Engl J Med 333, 333–336 (1995).
Leemans, J. C., Cassel, S. L. & Sutterwala, F. S. Sensing damage by the NLRP3 inflammasome. Immunol. Rev 243, 152–162 (2011).
Ciraci, C., Janczy, J. R., Sutterwala, F. S. & Cassel, S. L. Control of innate and adaptive immunity by the inflammasome. Microbes Infect 14, 1263–1270 (2012).
Anders, H. J. & Muruve, D. A. The inflammasomes in kidney disease. J Am Soc Nephrol 22, 1007–1018 (2011).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481, 278–286 (2012).
Latz, E., Xiao, T. S. & Stutz, A. Activation and regulation of the inflammasomes. Nat Rev Immunol 13, 397–411 (2013).
Iyer, S. S. et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA 106, 20388–20393 (2009).
Shigeoka, A. A. et al. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol 185, 6277–6285 (2010).
Kim, H. J. et al. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J Pharmacol Exp Ther 346, 465–472 (2013).
Vilaysane, A. et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol 21, 1732–1744 (2010).
Bakker, P. J. et al. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int 85, 1112–1122 (2014).
Hoffman, H. M., Mueller, J. L., Broide, D. H., Wanderer, A. A. & Kolodner, R. D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet 29, 301–305 (2001).
Hull, K. M., Shoham, N., Chae, J. J., Aksentijevich, I. & Kastner, D. L. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol 15, 61–69 (2003).
Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Verma, D. et al. The Q705K polymorphism in NLRP3 is a gain-of-function alteration leading to excessive interleukin-1beta and IL-18 production. PLoS One 7, e34977 (2012).
Roberts, R. L. et al. Evidence of interaction of CARD8 rs2043211 with NALP3 rs35829419 in Crohn’s disease. Genes Immun 11, 351–356 (2010).
Pontillo, A., Vendramin, A., Catamo, E., Fabris, A. & Crovella, S. The missense variation Q705K in CIAS1/NALP3/NLRP3 gene and an NLRP1 haplotype are associated with celiac disease. Am J Gastroenterol 106, 539–544 (2011).
Tan, M. S. et al. NLRP3 polymorphisms are associated with late-onset Alzheimer’s disease in Han Chinese. J Neuroimmunol 265, 91–95 (2013).
Villani, A. C. et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet 41, 71–76 (2009).
Haq, M., Norman, J., Saba, S. R., Ramirez, G. & Rabb, H. Role of IL-1 in renal ischemic reperfusion injury. J Am Soc Nephrol 9, 614–619 (1998).
Daemen, M. A., van’t Veer, C., Wolfs, T. G. & Buurman, W. A. Ischemia/reperfusion-induced IFN-gamma up-regulation: involvement of IL-12 and IL-18. J Immunol 162, 5506–5510 (1999).
Daemen, M. A. et al. Activated caspase-1 is not a central mediator of inflammation in the course of ischemia-reperfusion. Transplantation 71, 778–784 (2001).
Wang, W. et al. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J Immunol 190, 1239–1249 (2013).
Liu, D., Rhebergen, A. M. & Eisenbarth, S. C. Licensing Adaptive Immunity by NOD-Like Receptors. Front Immunol 4 (2013).
Ichinohe, T., Lee, H. K., Ogura, Y., Flavell, R. & Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med 206, 79–87 (2009).
Franchi, L. & Nunez, G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur. J. Immunol 38 (2008).
Kummer, J. A. et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J. Histochem. Cytochem 55, 443–452 (2007).
Solini, A. et al. The purinergic 2X7 receptor participates in renal inflammation and injury induced by high-fat diet: possible role of NLRP3 inflammasome activation. J. Pathol 231 (2013).
Homsi, E., Janino, P. & de Faria, J. B. Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int 69, 1385–1392 (2006).
Cavaille-Coll, M. et al. Summary of FDA workshop on ischemia reperfusion injury in kidney transplantation. Am J Transplant 13, 1134–1148 (2013).
Schroppel, B. & Legendre, C. Delayed kidney graft function: from mechanism to translation. Kidney Int (2014).
Roberts, R. L. et al. Interaction of the inflammasome genes CARD8 and NLRP3 in abdominal aortic aneurysms. Atherosclerosis 218, 123–126 (2011).
Paramel Varghese, G. et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J Am Heart Assoc 5 (2016).
Damman, J. et al. Association of complement C3 gene variants with renal transplant outcome of deceased cardiac dead donor kidneys. Am J Transplant 12, 660–668 (2011).
Dessing, M. C. et al. Toll-Like Receptor Family Polymorphisms Are Associated with Primary Renal Diseases but Not with Renal Outcomes Following Kidney Transplantation. PLoS. One 10, e0139769 (2015).
Acknowledgements
The authors would like to thank the members of the REGaTTA cohort (REnal GeneTics TrAnsplantation; University Medical Center Groningen, University of Groningen, Groningen, the Netherlands): H.G.D. Leuvink, H. van Goor, J.L. Hillebrands, B.G. Hepkema, H. Snieder, J. van den Born, M.H. de Borst, S.J.L. Bakker, G.J. Navis and M. Seelen. This study was supported by a grant of Dutch Kidney Foundation awarded to JCL and MCD (C06.6023) and an ERA-EDTA fellowship grant awarded to JK (69–201). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Study conception and design: J.D. and G.J.N. Acquisition of data: M.C.D., J.K. and J.D. Analysis and interpretation of data: M.C.D. and J.K. Drafting of manuscript: M.C.D., J.K., S.F. and J.C.L.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dessing, M., Kers, J., Damman, J. et al. Donor and recipient genetic variants in NLRP3 associate with early acute rejection following kidney transplantation. Sci Rep 6, 36315 (2016). https://doi.org/10.1038/srep36315
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36315
This article is cited by
-
The role of inflammasomes in human diseases and their potential as therapeutic targets
Signal Transduction and Targeted Therapy (2024)
-
Effects of intraoperative dexmedetomidine infusion on renal function in elective living donor kidney transplantation: a randomized controlled trial
Canadian Journal of Anesthesia/Journal canadien d'anesthésie (2022)
-
The role of inflammasomes in kidney disease
Nature Reviews Nephrology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
