
Abstract
Qa-1 epitopes, the peptides that bind to non-classical major histocompatibility complex Ib Qa-1 molecules and are recognized by Qa-1-restricted CD8+ regulatory T (Treg) cells, have been identified in pathogenic autoimmune cells that attack myelin sheath in experimental autoimmune encephalomyelitis (EAE, an animal model for multiple sclerosis [MS]). Additionally, immunization with such epitopes ameliorates the EAE. However, identification of such epitopes requires knowledge of the pathogenic autoimmune cells which are largely unknown in MS patients. Hence, we asked whether the CD8+ Treg cells could directly target the myelin sheath to ameliorate EAE. To address this question, we analyzed Qa-1 epitopes in myelin oligodendrocyte glycoprotein (MOG that is a protein in myelin sheath). Here, we report identification of a MOG-specific Qa-1 epitope. Immunization with this epitope suppressed ongoing EAE, which was abrogated by CD8+ T cell depletion. Additionally, the epitope immunization activated the epitope-specific CD8+ T cells which specifically accumulated in the CNS-draining cervical lymph nodes. Finally, CD8+ T cells primed by the epitope immunization transferred EAE suppression. Hence, this study reveals a novel regulatory mechanism mediated by the CD8+ Treg cells. We propose that immunization with myelin-specific HLA-E epitopes (human homologues of Qa-1 epitopes) is a promising therapy for MS.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS) is a chronic and debilitating disorder in the central nervous system (CNS). This disease is afflicting more than 2.5 million individuals worldwide. In addition, data suggest that MS global prevalence and incidence rate are increasing1. It is believed that the disease is caused by attacks on the myelin sheath by one’s own immune system (autoimmune attacks). Hence, current research efforts focus on developing strategies to arrest the autoimmune attacks. As a result, an array of medications has been approved by the FDA. These medications act to block either the functions of inflammatory molecules or the entrance of immune cells into the CNS2. Therefore, the medications do not specifically block the autoimmune attacks on the myelin sheath. Because the immune system uses the same mechanisms to attack the myelin sheath as those to combat health hazards (e.g. infections and cancers), current medications compromise the immune defense mechanism and are still complicated by severe side effects, particularly infections and cancers3,4.
Accordingly, the ultimate goal of MS therapy is to specifically arrest the autoimmune attacks on the myelin sheath, while sparing global immune defense mechanisms5. In principle, antigen-specific therapy is the logical pathway to achieve this goal5,6. In this regard, the major purpose of an antigen-specific therapy is to specifically instruct potentially pathogenic myelin-specific autoimmune cells, which are responsible for the EAE and MS7,8,9,10,11, to become myelin-specific regulatory T (Treg) cells. Such Treg cells can then specifically arrest the autoimmune attacks on the myelin sheath without compromising the immune defense mechanisms. However, there is currently no FDA-approved antigen-specific therapy for MS.
Among numerous antigen-specific therapies that are being investigated, the strategies that utilize regulatory Qa-1 epitopes to enhance the function of Qa-1-restricted CD8+ Treg cells have unique advantages. In this regard, Qa-1 epitopes are the peptides that bind to non-classical major histocompatibility complex (MHC) Ib Qa-1 molecules and are targets of the Qa-1-restricted CD8+ T cells. To support the importance of this Qa-1-epitope-CD8 axis in antigen-specific therapy of MS, recent data have convincingly demonstrated that the dominant role of Qa-1 molecules is presentation of regulatory Qa-1epitopes to the Qa-1-restricted CD8+ Treg cells12,13,14,15. Indeed, immunization with dendritic cells (DCs) pulsed with the Qa-1 epitopes, derived from pathogenic autoimmune cells, has been shown to specifically suppress EAE through down regulation of the pathogenic autoimmune cells16,17,18,19. These animal studies suggest that HLA-E epitopes (the human homologues of murine Qa-1 epitopes) derived from pathogenic autoimmune cells are promising therapeutic agents for MS. However, in MS patients, pathogenic autoimmune cells are largely unknown and hard to determine. Therefore, identification of appropriate HLA-E epitopes in the pathogenic autoimmune cells, if possible, is difficult.
Although pathogenic autoimmune cells have been intensively investigated as the targets of Qa-1-mediated antigen-specific therapy, myelin sheath (i.e. the tissue that is attacked by one’s own immune system in MS patients) has been the target of most antigen-specific therapies5. Therefore, we hypothesized that regulatory HLA-E epitopes, specifically located in the myelin sheath, were present and that immunization with such myelin-specific HLA-E epitopes activated the epitope-specific HLA-E-restricted CD8+ Treg cells to ameliorate MS. To test this hypothesis, we investigated potential Qa-1 epitopes (the murine homologues of human HLA-E epitopes) in myelin oligodendrocyte glycoprotein (MOG) that is one of the myelin proteins in myelin sheath. Additionally, we studied whether immunization with such epitopes could augment the function of the Qa-1-restricted CD8+ T cells to ameliorate EAE. The following is a detailed description of our findings from this study.
Results
Portion of CD8+ T cells in the CD8+ T cell lines reactive to the pool of OLPs (overlapping peptides) covering the whole length of mouse MOG is Qa-1b restricted
Current data suggest that Qa-1-restricted CD8+ Treg cells can target pathogenic autoimmune cells20 and suppress EAE, an animal model of human MS. In this case, the CD8+ T cells achieve the targeting by recognizing regulatory Qa-1 epitopes that are expressed in the myelin-specific pathogenic autoimmune cells16,19,21. However, these regulatory Qa-1 epitopes, though easily identified in animal models, are difficult to define in humans because pathogenic autoimmune cells by themselves are hard to determine in MS patients. This obstacle has prevented further clinical translation of the Qa-1-restricted CD8+ Treg cells.
Since pathogenic autoimmune cells in EAE and MS mainly attack myelin sheath, we asked whether Qa-1-restricted CD8+ Treg cells could specifically target myelin sheath as well. To answer this question, we proceeded to address if regulatory Qa-1b epitopes which are the targets of Qa-1-restricted CD8+ Treg cells were present in mouse MOG since MOG is one of the myelin proteins in myelin sheath. In order to map potential Qa-1b epitopes in mouse MOG, we generated a 15 mer OLP library that covered the whole length of mouse MOG (247 amino acids, or 247aa) from N- to C-termini. All of the OLPs in this library were 15aa in length and overlapped by 11aa. Hence the OLP library contained 59 OLPs in total (Fig. 1, panel A). A pool of the 59 OLPs (MOG_pool), which contained a final concentration of 4.2 μg/ml for each individual peptide, was used to stimulate CD8+ T cells purified from Kb−/−Db−/− mice for generating MOG_pool-reactive CD8+ T cell lines in vitro. Here, we utilized CD8+ T cells that were purified from Kb−/−Db−/− mice because CD8+ T cells in these mice were restricted mostly by non-classical MHC Ib molecules including the Qa-1b. During in vitro stimulations, CD8+ T cell lines were monitored weekly for response, using IFN-γ Enzyme-linked ImmunoSpot, to the MOG_pool in the presence of either C1R or C1R.Qa-1b cells. Our data showed that most CD8+ T cell lines we generated specifically responded to the MOG_pool in the presence of both C1R and C1R.Qa-1b cells (Fig. 1, panels B,C). However, response to the MOG_pool was much stronger when C1R.Qa-1b cells were present. The data therefore suggested that portion of the CD8+ T cells in the lines responded to the MOG_pool in a Qa-1b-dependent (or Qa-1-restricted) manner.

Portion of CD8+ T cells reactive to the pool of OLPs covering the whole length of mouse MOG was Qa-1b restricted.
(A) A schematic view of the mouse MOG OLP library. 15 mer peptides across the whole length of mouse MOG (247aa) and overlapped by 11aa were synthesized. A pool of the 59 OLPs (MOG_pool) at a concentration of 4.2 μg/ml for each peptide was then generated for stimulating CD8+ T cells purified from Kb−/−Db−/− mice. (B) Experimental design for “C”: CD8+ T cells purified from Kb−/−Db−/− mice were stimulated with the MOG_pool in vitro on a weekly basis. Before being stimulated each time, the CD8+ T cells were monitored for response to the MOG_pool, using IFN-γ Enzyme-linked ImmunoSpot, in the presence of either C1R or C1R.Qa-1b cells as antigen presenting cells. (C) Enzyme-linked ImmunoSpot data on day 7 were shown and were representative of three independent experiments. The number at upper left corner of each well is the absolute number of IFN-γ spot forming cells (SFCs) in the corresponding well (50,000 CD8+ T cells/well).
Recognition of multiple OLPs by the MOG_pool-reactive CD8+ T cell lines depends on Qa-1b
We next addressed which individual OLPs in the MOG_pool were recognized by the MOG_pool-reactive CD8+ T cell lines in a Qa-1b-restricted manner. Thus, the 59 individual OLPs were interrogated individually for their ability to stimulate the MOG_pool-reactive CD8+ T cell lines in the presence of either C1R or C1R.Qa-1b cells as antigen-presenting cells (Fig. 2). Data showed that most OLPs provided stronger stimulation of the MOG_pool-reactive CD8+ T cell lines in the presence of C1R.Qa-1b cells as compared to C1R cells (Fig. 2, panels B–D). However, only three OLPs, i.e. OLP68, OLP96, and OLP105, consistently stimulated the CD8+ T cell lines in a Qa-1b-restricted manner. We hence performed a detailed analysis of these three OLPs. Accordingly, Kb−/−Db−/− mice were immunized individually with the OLP68, OLP96, or OLP105. Ten days later, CD8+ T cells were purified from draining lymph nodes and stimulated with the corresponding peptides used for immunization. One week later, the CD8+ T cells were examined for response, using IFN-γ Enzyme-linked ImmunoSpot, to the corresponding peptides in the presence of either C1R or C1R.Qa-1b cells. Our data showed that all three OLPs stimulated CD8+ T cells in a Qa-1b-restricted manner (Fig. 2, panels E,F), suggesting that all three OLPs contained Qa-1b epitopes.

Recognition of multiple OLPs by the MOG_pool-reactive CD8+ T cell lines depended on Qa-1b.
(A–D) MOG_pool-reactive CD8+ T cell lines were generated as shown in Fig. 1. The 59 individual OLPs were interrogated, using IFN-γ Enzyme-linked ImmunoSpot, for their ability to stimulate the MOG_pool-reactive CD8+ T cell lines in the presence of either C1R or C1R.Qa-1b cells. “A” Showed the 96-well plate layout of the 59 individual OLPs. Numbers in the wells represented identification codes (IDs) for the 59 individual MOG OLPs, i.e. 49 ~ 107 from N to C termini. “B” and “C” Showed responses of one representative MOG_pool-reactive CD8+ T cell line to the 59 individual OLPs in the presence of either C1R (B) or C1R.Qa-1b (C) cells. Numbers represented SFCs (spot-forming cells)/106 CD8+ T cells. Numbers in “D” represented fold increases of SFCs/106 CD8+ T cells in the wells that contain C1R.Qa-1b cells (C) as compared to C1R cells (B), i.e. numbers = (SFCs per 106 CD8+ T cells in C)/(SFCs per 106 CD8 T cells in B). The highlighted were the top three OLPs that stimulated unequivocal Qa-1b-restricted CD8+ T cell response. (E) Experimental design for “F”: Kb−/−Db−/− mice were immunized with OLP68, OLP96, or OLP105. CD8 T cells purified ten days later were stimulated with the corresponding peptides used for immunization. After one week in vitro culture, the CD8 T cells were examined, using IFN-γ Enzyme-linked Immunospot, for specific responses to the corresponding peptides in the presence of either C1R or C1R.Qa-1b cells. (F) Representative data from two independent experiments were shown.
MOG196-204 (hereafter MOG196) is the minimal and optimal Qa-1b epitope in OLP105
Since OLP105 stimulated, in most assays, the highest number of SFCs (spot-forming cells)/106 CD8+ T cells in the MOG_pool-reactive CD8+ T cell lines in the presence of C1R.Qa-1b cells, we proceeded to further analyze the minimal and optimal Qa-1b epitope in this OLP. Thus, we synthesized progressively N- and C-terminally truncated peptides of OLP105 down to 6 mers because MHC I molecules could present epitopes of 7 ~ 10 mers. These truncated peptides and the original OLP105 were then examined for their ability to stimulate, using IFN-γ Enzyme-linked ImmunoSpot, an OLP105-reactive CD8+ T cell line in the presence of C1R.Qa-1b cells. Our data demonstrated that a 9 mer peptide, i.e. IICYNWLHR, was the minimal and optimal epitope in OLP105 (Fig. 3).

Fine mapping of the minimal and optimal Qa-1b epitope in OLP105.
Progressively N- and C-terminally truncated OLP105 peptides were synthesized and interrogated for their ability to stimulate, using IFN-γ Enzyme-linked ImmunoSpot, an OLP105-reactive CD8+ T cell line in the presence of C1R. Qa-1b cells. IDs and sequences of the truncated peptides were shown in the left and middle panels respectively. Corresponding bars in the right panel showed IFN-γ spot-forming cells (SFCs) per 106 CD8+ T cells in response to corresponding peptides. Highlighted bar and sequence displayed the highest response and stimulating peptide sequence respectively, demonstrating that sequence of the optimal and minimal epitope in OLP105 was IICYNWLHR, i.e. MOG196-204 (or MOG196). The data was representative of four independent experiments.
MOG196 binds to Qa-1b and activates MOG196-specific Qa-1b-restricted CD8+ T cells in vitro
To further confirm that MOG196 was a Qa-1b epitope, we next asked whether MOG196 could bind to Qa-1b by addressing whether this 9 mer peptide could successfully refold with the recombinant Qa-1b protein in vitro. Thus, MOG196 was incubated with recombinant Qa-1b protein and β2m at 10 °C for four days. The resulting solution was analyzed in a size exclusion column and displayed a distinct protein peak (Fig. 4, panel A, peak B), suggesting formation of MOG196/Qa-1b/β2m monomer. When the monomer was further analyzed in an anion exchange column, the monomer displayed two protein peaks (Fig. 4, panel B, peak B1 and B2). To further address potential reasons for the two protein peaks in the anion exchange column, proteins in peaks A, B1, and B2 were biotinylated. Portions of the biotinylated proteins were incubated with Streptavidin that was able to bind four biotinylated proteins to form tetramers. Subsequently, the biotinylated proteins and streptavidin-conjugated tetramers were analyzed in a non-denature protein gel. Data showed that biotinylated proteins in peak A did not show any distinct protein band (Fig. 4, panel C lane 1), supporting that this peak contained mainly non-specific protein aggregates. In contrast, biotinylated proteins in peak B1 displayed a single protein band (Fig. 4, panel C, lane 3), indicating correct formation of MOG196/Qa-1b/β2m monomer. Interestingly, biotinylated proteins in peak B2 exhibited two protein bands (Fig. 4, panel C, lane 5), suggesting that some proteins in this peak were not correctly refolded. Furthermore, addition of streptavidin successfully conjugated biotinylated proteins in peak B1 and B2 into tetramers (Fig. 4, panel C, lanes 4 and 6). Our data therefore demonstrated that MOG196 could bind to Qa-1b.

MOG196 could bind to Qa-1b and stimulate MOG196-specific Qa-1b-restricted CD8+ T cells.
(A) MOG196 was incubated with recombinant Qa-1b and β2 microglobulin (β2m) in a protein refolding buffer at 10 °C under gentle agitation (60 rpm) for four days. The solution was separated by a size exclusion column. Peak “A” and “B” represented typical non-specific protein aggregates and correctly refolded MOG196/Qa-1b monomer respectively. (B) Monomers in peak B of panel “A” were further analyzed by an anion exchange chromatography. (C) Proteins in peaks “A”, “B1”, and “B2” were biotinylated and portions of the biotinylated proteins were incubated with streptavidin (SA) to examine formation of tetramers. The biotinylated proteins and corresponding tetramers were analyzed in a non-denature protein gel. Arrows show protein bands for MOG196/Qa-1b monomers. “−”: without SA; “+”: with SA; “pk A”: peak A; “pk B1”: peak B1; “pk B2”: peak B2; “Std”: a protein standard. (D) CD8+ T cells were purified from naïve C57BL/6 (B6) mice, individually stimulated weekly with untreated (No peptide) or MOG196-pulsed (MOG196), either B6 (upper two plots) or Kb−/−Db−/− (lower two plots) macrophages. The CD8+ T cells were analyzed for binding to Qa-1b/MOG196 tetramer at days 0, 7, and 14. One representative data on day 14 from four individual experiments was shown.
To ask whether MOG196 could be presented by antigen-presenting cells to activate MOG196-specific Qa-1b-restricted CD8+ T cells, CD8+ T cells purified from C57BL/6 mice were stimulated in vitro weekly by MOG196-pulsed antigen-presenting cells derived from either Kb−/−Db−/− or C57BL/6 mice. Data demonstrated that, beginning on day 14 after in vitro re-stimulation, MOG196/Qa-1b tetramer+ cells could be detected in the CD8+ T cell lines (Fig. 4, panel D), indicating successful activation of MOG196-specific Qa-1b-restricted CD8+ T cells. In addition, antigen-presenting cells derived from both Kb−/−Db−/− and C57BL/6 mice were able to present MOG196 to activate the MOG196-specific Qa-1b-restricted CD8+ T cells in vitro (Fig. 4, panel D).
Immunization with MOG196-pulsed DCs ameliorates MOG35-55 EAE
To address whether MOG196 is a biologically relevant regulatory Qa-1b epitope, we first asked if immunization with the epitope-pulsed DCs ameliorated MOG35-55 EAE. Thus, we immunized C57BL/6 (B6) mice with MOG196-pulsed Kb−/−Db−/− DCs one week before and after the EAE induction. Our data clearly showed that immunization with the MOG196- but not the Qdm-pulsed Kb−/−Db−/− DCs significantly ameliorated the paralytic disease (Fig. 5A,B, Table S1). However, one might have questioned whether immunization with MOG196-pulsed wild-type DCs was also able to ameliorate EAE. To evaluate this potential, we immunized C57BL/6 mice with MOG196-pulsed B6 DCs on days -3, 2, and 7. On day 0, mice were immunized with MOG35-55 for inducing EAE. Our data again showed that immunization with wild-type DCs pulsed with MOG196, but not Qdm or HSP60p216, significantly ameliorated the paralytic disease (Fig. 5C,D, Table S2).

Immunization with MOG196-pulsed DCs suppressed MOG35-55-induced EAE.
(A) Experimental design for “B”: C57BL/6 mice were immunized with MOG35-55 for EAE. One week before and after the EAE induction, the animals received one of the following subcutaneous treatments: (1) no treatment (No Tx); (2) 1 × 106 Qdm-pulsed Kb−/−Db−/− DCs (DC/Qdm); (3) 1 × 106 MOG196-pulsed Kb−/−Db−/− DCs (DC/MOG196). The mice were then monitored for paralytic disease daily. (B) Daily mean disease score was shown. N = 5. *P < 0.05; **P < 0.01; ***P < 0.001. Two-way ANOVA test. (C) Experimental design for “D”: C57BL/6 mice were immunized with MOG35-55 for EAE on day 0. At days -3, 2, and 7, animals were immunized with 1 × 106 B6 DCs pulsed with Qdm (DC/Qdm), HSP60p216 (DC/HSP60p216), or MOG196 (DC/MOG196). The animals were then monitored for paralytic disease daily. (D) Daily mean disease score was shown. N = 5. *P < 0.05. Two-way ANOVA test. Concentration of peptides used for pulsing the DCs was 10 μg/ml.
Immunization with the MOG196-pulsed DCs suppresses ongoing MOG35-55 EAE, which is dependent on CD8+ T cells
Next, we asked whether immunization with the MOG196-pulsed mature B6 DCs could suppress ongoing EAE and whether the disease suppression depended on CD8+ T cells. To address the role of the CD8+ T cells, we decided to utilize a depleting anti-CD8 monoclonal antibody, i.e. the clone 53–6.7 that had been shown to specifically abrogate the function of CD8+ T cells both in vitro and in vivo16,22. Hence, C57BL/6 mice were immunized with the MOG35-55 for EAE. Ten days later, animals received either no treatment (No Tx) or the MOG196-pulsed DCs. Among the animals that received the MOG196-pulsed DCs, some animals also received an intra-peritoneal injection of the depleting anti-CD8 antibody at days -2, -1, 7 and 14 to deplete CD8+ T cells. Our data showed that, as compared to no treatment, one intravenous injection of 5 × 105 MOG196-pulsed B6 DCs robustly suppressed the ongoing paralytic disease (Fig. 6A,B, Table S3). Importantly, depletion of CD8+ T cells abrogated the protective effect of the DC/MOG196. Therefore, our data demonstrated that suppression of ongoing EAE by the MOG196-pulsed DC was dependent on CD8+ T cells.

Immunization with MOG196-pulsed DCs suppressed ongoing MOG35-55-induced EAE, which was dependent on CD8+ T cells.
(A) Experimental design for “B”: C57BL/6 mice were immunized with MOG55-55 for EAE on day 0. In one group, the animals were intraperitoneally injected with a monoclonal depleting anti-CD8 antibody (mAb) at days -2, -1, 7, and 14. At day 10, animals received either no treatment (No Tx) or one intravenous injection of mitomycin C-treated C57BL/6 DCs pulsed with MOG196 (DC/MOG196). Paralytic disease was monitored daily. (B) Daily mean disease score was shown. N = 5 (one animal that died from EAE before antibody treatment was excluded from this analysis). *P < 0.05; **P < 0.01; ***P < 0.001. Two-way ANOVA test. Data shown were representative of two independent experiments.
Immunization with the MOG196-pulsed DCs activates CD8+ T cells that transfer EAE suppression
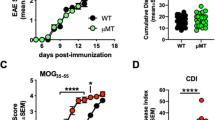
To determine whether function of the CD8+ Treg cells was augmented by the MOG196 immunization, CD8+ T cell donor C57BL/6 mice intravenously received no immunization (none-immune), DCs (DC), or MOG196-pulsed DCs (MOG196-DC). Twenty days later, CD8+ T cells were purified from the donor animals and transferred into new host C57BL/6 mice that were immunized with the MOG35-55 for inducing EAE on the second day. The animals were monitored for the paralytic symptoms daily (Fig. 7A). Our data showed that transfer of the CD8+ T cells from animals immunized with the MOG196-pulsed DCs but not the control CD8+ T cells significantly suppressed the EAE (Fig. 7B).

Immunization with the MOG196-pulsed DCs activates CD8+ T cells that transfer EAE suppression.
(A) C57BL/6 mice intravenously received no immunization (none-immune), DCs (DC), or MOG196-pulsed DCs (MOG196-DC). Twenty days later, CD8+ T cells from the animals in each group were purified and pooled. Host animals were divided into the following groups: no treatment; DC/CD8 in which each animal received 1 × 106 CD8+ T cells from the animals immunized with the DCs; MOG196-DC/CD8 in which each animal received 1 × 106 CD8+ T cells from the animals immunized with the MOG196-pulsed DCs; or none-immune/CD8 in which each animal received 1 × 106 CD8+ T cells from the none-immunized animals. On the second day, all animals were immunized with the MOG35-55 for inducing EAE and monitored for paralytic symptoms daily. (B) Cumulative data of mean disease scores from five animals in each group. *P < 0.05 No Tx vs. MOG196-DC/CD8. Mann Whitney test.
Immunization with MOG196-pulsed DCs activates Qa-1b/MOG196 tetramer+ cells that accumulate in the cervical lymph nodes in EAE-bearing animals
Next, we asked whether immunization with MOG196-pulsed DCs indeed activated Qa-1b/MOG196 tetramer+ cells in vivo in EAE-bearing animals. Hence, C57BL/6 mice were induced for EAE. Ten days later, when the paralytic disease began, animals received mature DC2.4 cells (DC2.4 is a bone-marrow-derived DC line)23 or MOG196-pulsed DC2.4 cells (DC2.4/MOG196). Four days after the treatments, spleens and cervical lymph nodes were analyzed for the presence of Qa-1b/MOG196 tetramer+ cells. Our data showed that percent of Qa-1b/MOG196 tetramer+ cells in spleens between the two treatments was similar, while percent of tetramer+ cells in cervical lymph nodes following the DC2.4/MOG196 treatment, as compared to DC2.4 treatment, was significantly elevated (Fig. 8A–C). The data suggested that the Qa-1b/MOG196 tetramer+ cells specifically accumulated in the CNS in EAE-bearing animals. In contrast, I-Ab/MOG38-49 tetramer+ cells, which detected MOG35-55-reactive pathogenic autoimmune cells, were significantly reduced in cervical lymph nodes (Fig. 8A,D,E). The data were consistent with suppression of ongoing paralytic disease following the treatment with the MOG196-pulsed DCs (Fig. 6).

Immunization with DCs pulsed with MOG196 activates Qa-1b/MOG196 tetramer+ cells that specifically accumulate in cervical lymph nodes.
(A) C57BL/6 mice (5 mice/group) were immunized with MOG35-55 for EAE. Ten days later, when paralytic symptoms began, animals received one intravenous immunization with mytomycin C-treated DC2.4 or MOG196-pulsed DC2.4 (DC2.4/MOG196). Four days later, mononuclear cells prepared from spleens and cervical lymph nodes were examined for the presence of Qa-1b/MOG196 and I-Ab/MOG38-49 tetramer+ cells by flow cytometry. (B) Representative plots of Qa-1b/MOG196 tetramer+ cells among CD8+ T cells in cervical lymph nodes and spleens. (C) Cumulative data of Qa-1b/MOG196 tetramer+ cells from five mice. *P < 0.05. Two-way ANOVA test. (D) Representative plots of I-Ab/MOG38-49 tetramer+ cells among CD8− T cells in cervical lymph nodes and spleens. (E) Cumulative data of I-Ab/MOG38-49 tetramer+ cells from five mice. *P < 0.05. Two-way ANOVA test. (F) Experimental design for “G”: C57BL/6 mice were intravenously immunized with MOG196-pulsed DC2.4 cells at days 0 and 10. Ten days after the last immunization, mononuclear cells from cervical lymph nodes and spleens were examined for the expressions of CD122 and Ly49 on Qa-1b/MOG196 tetramer+ CD8+ T cells by flow cytometry. (G) Representative FACS plots from two independent experiments were shown.
CD122 and Ly49 are expressed in a portion of the Qa-1b/MOG196 tetramer+ cells
Recent data suggest that Qa-1-restricted CD8+ Treg cells are among CD122+Ly49+ CD8+ T cells12,24,25. We hence analyzed expression of these two markers on the Qa-1b/MOG196 tetramer+ cells. Our data showed that about 22.6% of the tetramer+ cells expressed CD122 and about 7.2% of the tetramer+ cells expressed both CD122 and Ly49 (Fig. 7F,G). Therefore, the data suggest that CD122 and Ly49 are two important markers for Qa-1-restricted CD8+ Treg cells. However, additional markers are needed to more precisely identify this subset of CD8+ Treg cells.
MOG196 is evolutionarily conversed and unique to MOG
Some known Qa-1-binding peptides, e.g. Qdm and HSP60p216, bind to both Qa-1 and HLA-E26. In addition, peptide binding motifs of Qa-1 and HLA-E are similar26,27. We therefore asked whether MOG196 sequence was evolutionarily conserved. Accordingly, blast search of Genbank protein sequence data showed that MOG196 sequence was evolutionarily conserved (Fig. S1). Furthermore, sequence of MOG196 is specific to MOG and located in the intracellular domain (Fig. S2).
Discussion
In this article, for the first time, we present evidence for the existence of a regulatory Qa-1b epitope (i.e. MOG196-204 or MOG196) in myelin oligodendrocyte glycoprotein (MOG) that is a myelin protein in myelin sheath. Immunization with the epitope-pulsed dendritic cells (DCs) suppresses ongoing EAE and activates CD8+ T cells that specifically accumulate in cervical lymph nodes. Additionally, CD8+ T cells in vivo primed by the MOG196-pulsed DCs transfer EAE suppression. Hence, this study opens a potential new avenue for the antigen-specific therapy of multiple sclerosis (MS).
The major purpose of antigen-specific therapy is to immunize an individual with pathogenic myelin epitopes (i.e. the myelin epitopes in the myelin sheath targeted by the pathogenic autoimmune cells) under a tolerogenic condition. Such immunization specifically instructs the potentially pathogenic myelin-specific autoimmune cells, which cause the EAE and MS7,8,9,10,11, to become myelin-specific regulatory T (Treg) cells. These Treg cells can then specifically arrest the autoimmune attacks on the myelin sheath without compromising the immune defense mechanisms. In this regard, one such antigen-specific therapy is the tolerogenic dendritic cell (DC) that is generated in vitro under a tolerogenic condition. When pulsed with the pathogenic myelin epitopes, the tolerogenic DCs can induce the epitope-specific Treg cells28,29,30. However, recent data suggest that the tolerogenic DC is unstable in an in vivo pro-inflammatory environment and can be converted into a disease-worsening immunogenic DC31,32,33. This instability is hampering clinical translation of the tolerogenic DC31,34,35,36,37,38,39. In contrast, we show here that immunogenic DCs can be used to augment the MOG196-specific CD8+ Treg cells. Therefore, the MOG196 therapy does not have the instability concern. An additional advantage is that, unlike the previously reported antigen-specific therapies, the therapeutic strategy described in this report does not depend on the knowledge of the pathogenic myelin epitopes which are still under debate in MS patients.
Although this is the first report on the presence of regulatory Qa-1 epitopes in myelin proteins, previous studies have indicated that immunization with regulatory Qa-1(HLA-E) epitopes is indeed a logical approach for the treatment of EAE. First, animals that were deficient in Qa-1 displayed exaggerated secondary CD4+ T cell responses13. Second, Qa-1-restricted CD8+ T cells in animals whose Qa-1 molecules could not interact with inhibitory NKG2A molecules (Qa-1/R72A mutant mice), as compared with those in wild type animals, showed much stronger suppressive activities14. Third, aged animals whose Qa-1 molecules could not interact with CD8 molecules (Qa-1/D227K mutant mice) developed lupus-like autoimmune phenomena12. Hence, Qa-1-restricted CD8+ T cells are promising sources for antigen-specific therapy.
In addition to the above referred evidence suggesting a dominant regulatory role of Qa-1-restricted CD8+ T cells, regulatory Qa-1 epitopes in pathogenic autoimmune CD4+ T cells have been described and immunization with these epitopes has been shown to prevent EAE induction. Such epitopes include the p42–50 (GLRLIHYSY) located in the T cell receptor (TCR) Vβ8.2 protein16,17,18 and the LLSWVALFL peptide located in the TCRVβ8.1 protein21. Additionally, immunization with the HSP60sp (QMRPVSRAL) located in the leader sequence of the 60 Kda heat shock protein (HSP60) has been shown to prevent the progression of type 1 diabetes in NOD mice19. Finally, immunization with the HSP60p216 (GMKFDRGYI) located in the HSP60 has been shown to suppress ongoing collagen-induced arthritis (CIA), suggesting therapeutic potential of the epitope-specific, Qa-1-restricted CD8+ Treg cells25. All these previously identified Qa-1 epitopes are located in the pathogenic autoimmune cells. It has been proposed that immunization with these epitopes activates Qa-1-restricted CD8+ Treg cells that target the pathogenic autoimmune cells to ameliorate autoimmune diseases (e.g. EAE, CIA, and T1D)19,21,25,40,41. Because the identity of the pathogenic autoimmune cells is hard to determine in patients with MS42, clinical translation of these regulatory Qa-1 epitopes is difficult.
As mentioned above, previous studies of Qa-1(HLA-E)-mediated antigen-specific therapy were focused on identification of the regulatory Qa-1 epitopes in the pathogenic autoimmune CD4+ T cells. Since efficient entering of CD8+ T cells into CNS depends on presentation of CNS-antigens at the blood-brain barrier (BBB)43, Qa-1-restricted CD8+ Treg cells which target epitopes located in the pathogenic autoimmune CD4+ T cells may not efficiently accumulate in the CNS due to a lack of specificity for CNS and therefore may predominantly suppress the pathogenic CD4+ T cells in the peripheral lymphoid organs. In contrast, myelin-specific Qa-1-restricted CD8+ Treg cells described here will have the advantage to cross the BBB and provide in situ suppression of demyelinating inflammation in the CNS. To support this notion, our data showed that, in the course of EAE, MOG196-specific, Qa-1-restricted CD8+ Treg cells specifically accumulated in the cervical lymph nodes (i.e. the draining lymph nodes for CNS).
With respect to the potential mechanisms underlying the disease suppression mediated by the myelin-specific, Qa-1(HLA-E)-restricted CD8+ Treg cells, previous data suggest that the disease suppression involves IFN-γ, perforin, and IL-1512,44. These data appear to support a direct killing of target cells by the Qa-1-restricted CD8+ Treg cells. Since oligodendrocytes normally do not express MHC molecules45, it may explain that the MOG196-specific Qa-1-restricted CD8+ Treg cells do not damage myelin. We propose that the same antigen presenting cell, which phagocytoses myelin, can present both pathogenic and regulatory myelin epitopes to pathogenic CD4+ and regulatory CD8+ T cells respectively because both epitopes are located in the myelin (Fig. S3). In this regard, recognition of the regulatory Qa-1/HLA-E myelin epitopes by the CD8+ Treg cells leads to (1) elimination of and/or induction of tolerance in the antigen-presenting cells; (2) elimination and/or induction of tolerance in encephalitogenic T cells that have obtained the epitope complexes from antigen-presenting cells via a process called trogocytosis46,47,48. Consequently, activation and proliferation of encephalitogenic T cells are thwarted and autoimmune attacks of myelin sheath are stopped.
A similar recent finding showed that neuroantigen-specific CD8+ T cells were regulatory. In this regard, our myelin-specific Qa-1-restricted CD8+ Treg cells bear certain similarity to this recently reported neuroantigen-specific CD8+ Treg cell subset in terms of their shared specificity to myelin proteins and their dependency on IFN-γ and Perforin12,44,49. However, the neuroantigen-specific CD8+ Treg cells differ from myelin-specific Qa-1-restricted CD8+ Treg cells in requiring classical MHC I molecules for presentation50. In addition, priming of the neuroantigen-specific CD8+ Treg cells requires the presence of both the regulatory and pathogenic epitopes in the same myelin protein50. This second characteristic suggests that active immunization with such a regulatory neuroantigenic epitope is not a suitable therapy for MS. In contrast, the myelin-specific CD8+ Treg cells described here can be primed through active immunization with DCs pulsed with the regulatory epitope alone.
In addition to the neuroantigen-specific CD8+ Treg cells, it was shown that glatiramer, an FDA-approved first line medication for MS and a peptide mimicking myelin basic protein51, induced HLA-E-restricted CD8+ Treg cells in humans52. In a MS animal model, glatiramer-mediated suppression of EAE required CD8+ T cells and non-classical MIH Ib molecules53. Therefore, although glatiramer has multiple functions54, induction of Qa-1(HLA-E)-restricted CD8+ Treg cells may partially contribute to its therapeutic effect for EAE and MS. Despite glatiramer mimicking myelin basic protein, it has not been demonstrated that it is a strictly myelin-specific antigen. Hence, to our knowledge, the epitope described here is the first myelin-specific regulatory Qa-1 epitope that can actively and independently elicit myelin-specific CD8+ Treg cells.
Furthermore, it has been shown that immunization with a myelin epitope via anterior chamber (AC) of the eye (intracameral injection) induces the epitope-specific CD8+ Treg cells in the peripheral lymphoid tissues55,56. Interestingly, suppressive activity of such CD8+ Treg cells requires compatibility of the Qa-1 haplotype between the CD8+ Treg cells and the target cells57, suggesting that Qa-1 molecules are necessary for the CD8+ Treg cells to recognize the targets. However, the mechanisms by which the Qa-1 molecules are involved in the induction of such CD8+ Treg cells needs further investigation.
Although the myelin-specific Qa-1(HLA-E) epitopes are critical for priming the Qa-1(HLA-E)-restricted CD8+ Treg cells which bear the specificity for the myelin sheath, recent data suggest that other strategies may facilitate expansion of these CD8+ Treg cells. One potential such agent is the granulocyte macrophage colony stimulating factor (GM-CSF) that has been shown to stimulate the expression of OX40L and Jagged-1 in DCs which promote proliferation of Treg cells58,59. It would be interesting to know whether the GM-CSF-differentiated DCs, when pulsed with the myelin-specific Qa-1(HLA-E) epitope, can indeed augment expansion of the epitope-specific Qa-1(HLA-E)-restricted CD8+ Treg cells.
Methods
Mice, cell lines, and reagents
Mice
C57BL/6 (B6, female, 8–10 weeks of age, 18–20 g) and Kb−/−Db−/− mice were obtained from Taconic Farms and The Jackson Laboratory (Bar Harbor, Maine, USA) housed in a specific pathogen-free animal facility at the University of Texas at El Paso (UTEP) or Loma Linda University (LLU). All experiments were done in compliance with an Institutional Animal Care and Use Protocol approved by UTEP and/or LLU Animal Care and Use Committee.
Cell lines
C1R is a human B lymphoblastoid cell line. C1R.Qa-1b is a stable transfectant that constitutively expresses Qa-1b. DC2.4 is a DC line generated from bone-marrow-derived DCs and was kindly provided by Dr. Kenneth L. Rock23.
Reagents
Peptides used in this study were synthesized at Genemed Synthesis, Inc., San Antonio, TX 78244.
IFN-γ Enzyme-linked ImmunoSpot assay
Briefly, plates were coated with an anti-mouse IFN-γ mAb (5 μg/ml in PBS) at 4 °C overnight, blocked with culture medium for two hours at room temperature, and added with desired numbers of CD8+ T cells, peptides, and irradiated antigen-presenting cells. After cultured at 37 °C and 5% CO2 for 16 hours, plates were incubated with a biotinylated anti-mouse IFN-γ mAb (2 μg/ml) for two hours followed by streptavidin-conjugated HRP (horseradish peroxidase) for one hour. Finally, plates were incubated with a substrate, monitored for spot development, and washed with distilled water when spots were fully developed. After air-dried, plates were analyzed on an Enzyme-linked ImmunoSpot plate reader for enumerating spots in each well.
In vitro refolding of a peptide with recombinant Qa-1b protein
Refolding of MOG196 with recombinant Qa-1b protein was performed in the NIH tetramer core facility (Emory University, Atlanta, GA)60.
Induction of MOG35-55 -induced EAE
C57BL/6 mice were immunized subcutaneously with either 200 μg MOG35–55 emulsified in Incomplete Freund Adjuvant (IFA) supplemented with 250 μg heat-inactivated mycobacterium tuberculosis H37Ra (Difco Laboratories, Michigan, USA). On day 0 and 2, each mouse was administered 150 ng pertussis toxin (Calbiochem, Germany) intraperitoneally. Animals were then assessed for paralytic disease daily using the following scale: “0” = no paralysis, “1” = limp tail, “2” = limp tail and weak gait, “3” = hind limb paralysis, “4” = fore limb paralysis (animals were euthanized at or beyond stage “4”).
Generation of bone-marrow-derived DCs
Briefly, bone marrow single cell suspensions (1 × 106 cells/ml) containing 10 U/ml IL-4 and 100 U/ml GM-CSF were seeded into a 6-well plate (4 ml/well) and cultured at 37 °C, 5% CO2. On day 2, after non-adherent cells were carefully removed, plates were replenished with fresh media and cytokines. On day 4, non-adherent cells containing fresh media and cytokines were transferred into a new 6-well plate. On day 6, cells were replenished with fresh media and cytokines and stimulated with 0.1 μg/ml LPS overnight. On day 7, cells were collected for experiments.
Mytomycin C treatment of DCs and peptide pulsing
LPS activated DCs at the concentration of 5 × 107 cells/ml in PBS were treated by mytomycin C (50 μg/ml) for 20 minutes at 37 °C and 5% CO2 (DC2.4 cells were treated for 30 minutes). The treated DCs were then washed three times and adjusted to 5 × 106 cells/ml in serum-free medium containing 100 μg/ml MOG196 or HSP60P216 peptide. The cells were pulsed with the peptides for three to four hours at room temperature. After pulsing, the cells were washed once, reconstituted in PBS, and intravenously (or subcutaneously) injected into animals at 200 μl/mouse (5 × 105 cells or 1 × 106 cells/mouse).
Multichromatic flow cytometry
Briefly, about 0.5 ~ 1 × 106 cells in a FACS buffer (PBS containing 1% FBS and 0.05% sodium azide) were stained with various fluorescence-conjugated antibodies specific for the desired cell surface proteins or with tetramers at 4 °C for 30 min. The stained cells were washed twice in the FACS buffer before being analyzed on a BD FACSAria II.
Statistical analysis
All the statistical analyses were performed using One-Way or Two-Way ANOVA, followed by the SNK-q test or the Dunnett’s Multiple Comparison test. A P-value less than 0.05 was considered statistically significant.
Additional Information
How to cite this article: Wang, X. et al. Targeting Non-classical Myelin Epitopes to Treat Experimental Autoimmune Encephalomyelitis. Sci. Rep. 6, 36064; doi: 10.1038/srep36064 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Melcon, M. O., Correale, J. & Melcon, C. M. Is it time for a new global classification of multiple sclerosis? Journal of the neurological sciences 344, 171, doi: 10.1016/j.jns (2014).
Ben-Nun, A. et al. From classic to spontaneous and humanized models of multiple sclerosis: Impact on understanding pathogenesis and drug development. J Autoimmun, doi: 10.1016/j.jaut (2014).
Clifford, D. B. et al. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. The Lancet. Neurology 9, 438, doi: 10.1016/S1474-4422(10)70028-4 (2010).
Linda, H. et al. Progressive multifocal leukoencephalopathy after natalizumab monotherapy. The New England journal of medicine 361, 1081, doi: 10.1056/NEJM (2009).
Steinman, L. The re-emergence of antigen-specific tolerance as a potential therapy for MS. Mult Scler 21, 1223, doi: 10.1177/13524585155814411352458515581441 (2015).
Lutterotti, A., Sospedra, M. & Martin, R. Antigen-specific therapies in MS - Current concepts and novel approaches. Journal of the neurological sciences 274, 18, doi: 10.1016/j.jns (2008).
Hersh, C. M. & Cohen, J. A. Alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Immunotherapy 6, 249, doi: 10.2217/imt (2014).
McNally, J. P. et al. Eliminating encephalitogenic T cells without undermining protective immunity. J Immunol 192, 73, doi: 10.4049/jimmunol (2014).
Palanichamy, A. et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol 193, 580, doi: 10.4049/jimmunol (2014).
Kozovska, M. E. et al. Interferon beta induces T-helper 2 immune deviation in MS. Neurology 53, 1692 (1999).
Miller, A. et al. Treatment of multiple sclerosis with copolymer-1 (Copaxone): implicating mechanisms of Th1 to Th2/Th3 immune-deviation. J Neuroimmunol 92, 113 (1998).
Kim, H. J., Verbinnen, B., Tang, X., Lu, L. & Cantor, H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature 467, 328, doi: 10.1038/nature (2010).
Hu, D. et al. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat Immunol 5, 516 (2004).
Lu, L., Kim, H. J., Werneck, M. B. & Cantor, H. Regulation of CD8+ regulatory T cells: Interruption of the NKG2A-Qa-1 interaction allows robust suppressive activity and resolution of autoimmune disease. Proc Natl Acad Sci USA 105, 19420 (2008).
Jiang, H., Braunstein, N. S., Yu, B., Winchester, R. & Chess, L. CD8+ T cells control the TH phenotype of MBP-reactive CD4+ T cells in EAE mice. Proc Natl Acad Sci USA 98, 6301 (2001).
Tang, X. et al. Regulation of immunity by a novel population of Qa-1-restricted CD8alphaalpha+TCRalphabeta+ T cells. J Immunol 177, 7645, doi: 177/11/7645 (2006).
Tang, X., Maricic, I. & Kumar, V. Anti-TCR antibody treatment activates a novel population of nonintestinal CD8 alpha alpha+ TCR alpha beta+ regulatory T cells and prevents experimental autoimmune encephalomyelitis. J Immunol 178, 6043, doi: 178/10/6043 (2007).
Smith, T. R. et al. Dendritic cells use endocytic pathway for cross-priming class Ib MHC-restricted CD8alphaalpha+TCRalphabeta+ T cells with regulatory properties. J Immunol 182, 6959 (2009).
Wu, Y., Zheng, Z., Jiang, Y., Chess, L. & Jiang, H. The specificity of T cell regulation that enables self-nonself discrimination in the periphery. Proc Natl Acad Sci USA 106, 534 (2009).
Jiang, H. et al. Murine CD8+ T cells that specifically delete autologous CD4+ T cells expressing V beta 8 TCR: a role of the Qa-1 molecule. Immunity 2, 185 (1995).
Panoutsakopoulou, V. et al. Suppression of autoimmune disease after vaccination with autoreactive T cells that express Qa-1 peptide complexes. J Clin Invest 113, 1218 (2004).
Jiang, H., Zhang, S. I. & Pernis, B. Role of CD8+ T cells in murine experimental allergic encephalomyelitis. Science 256, 1213 (1992).
Shen, Z., Reznikoff, G., Dranoff, G. & Rock, K. L. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol 158, 2723 (1997).
Kim, H. J. et al. CD8+ T regulatory cells express the Ly49 Class I MHC receptor and are defective in autoimmune prone B6-Yaa mice. Proc Natl Acad Sci USA 108, 2010, doi: 10.1073/pnas (2011).
Leavenworth, J. W., Tang, X., Kim, H. J., Wang, X. & Cantor, H. Amelioration of arthritis through mobilization of peptide-specific CD8+ regulatory T cells. The Journal of clinical investigation 123, 1382, doi: 10.1172/JCI (2013).
Miller, J. D. et al. Analysis of HLA-E peptide-binding specificity and contact residues in bound peptide required for recognition by CD94/NKG2. J Immunol 171, 1369 (2003).
Kurepa, Z. & Forman, J. Peptide binding to the class Ib molecule, Qa-1b. J Immunol 158, 3244 (1997).
Yeste, A., Nadeau, M., Burns, E. J., Weiner, H. L. & Quintana, F. J. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 109, 11270, doi: 10.1073/pnas (2012).
Gross, C. C., Jonuleit, H. & Wiendl, H. Fulfilling the dream: tolerogenic dendritic cells to treat multiple sclerosis. Eur J Immunol 42, 569, doi: 10.1002/eji (2012).
Ilarregui, J. M. et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat Immunol 10, 981, doi: 10.1038/ni (2009).
Raker, V. K., Domogalla, M. P. & Steinbrink, K. Tolerogenic Dendritic Cells for Regulatory T Cell Induction in Man. Front Immunol 6, 569, doi: 10.3389/fimmu (2015).
Lim, D. S., Kang, M. S., Jeong, J. A. & Bae, Y. S. Semi-mature DC are immunogenic and not tolerogenic when inoculated at a high dose in collagen-induced arthritis mice. Eur J Immunol 39, 1334, doi: 10.1002/eji (2009).
Voigtlander, C. et al. Dendritic cells matured with TNF can be further activated in vitro and after subcutaneous injection in vivo which converts their tolerogenicity into immunogenicity. J Immunother 29, 407, doi: 10.1097/01.cji (2006).
Zhou, X. et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol 10, 1000, doi: 10.1038/ni (2009).
Schmitt, E. G. et al. IL-10 produced by induced regulatory T cells (iTregs) controls colitis and pathogenic ex-iTregs during immunotherapy. J Immunol 189, 5638, doi: 10.4049/jimmunol (2012).
Xu, L., Kitani, A., Fuss, I. & Strober, W. Cutting edge: regulatory T cells induce CD4 + CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol 178, 6725 (2007).
Murai, M. et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol 10, 1178, doi: 10.1038/ni (2009).
Tsuji, M. et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science 323, 1488, doi: 10.1126/science (2009).
Hardenberg, G., Steiner, T. S. & Levings, M. K. Environmental influences on T regulatory cells in inflammatory bowel disease. Semin Immunol 23, 130, doi: 10.1016/j.smim (2011).
Smith, T. R. & Kumar, V. Revival of CD8+ Treg-mediated suppression. Trends Immunol 29, 337, doi: 10.1016/j.it (2008).
Tang, X. L., Smith, T. R. & Kumar, V. Specific control of immunity by regulatory CD8 T cells. Cell Mol Immunol 2, 11 (2005).
Lehmann, P. V., Forsthuber, T., Miller, A. & Sercarz, E. E. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 358, 155, doi: 10.1038/358155a0 (1992).
Galea, I. et al. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med 204, 2023, doi: jem.20070064 (2007).
Beeston, T., Smith, T. R., Maricic, I., Tang, X. & Kumar, V. Involvement of IFN-gamma and perforin, but not Fas/FasL interactions in regulatory T cell-mediated suppression of experimental autoimmune encephalomyelitis. J Neuroimmunol 229, 91, doi: 10.1016/j.jneuroim (2010).
Goverman, J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol 9, 393, doi: 10.1038/nri (2009).
Umeshappa, C. S. et al. CD4+ Th-APC with acquired peptide/MHC class I and II complexes stimulate type 1 helper CD4+ and central memory CD8+ T cell responses. J Immunol 182, 193, doi: 182/1/193 (2009).
Joly, E. & Hudrisier, D. What is trogocytosis and what is its purpose? Nat Immunol 4, 815, doi: 10.1038/ni (2003).
Campana, S., De Pasquale, C., Carrega, P., Ferlazzo, G. & Bonaccorsi, I. Cross-dressing: an alternative mechanism for antigen presentation. Immunol Lett 168, 349, doi: 10.1016/j.imlet (2015).
Ortega, S. B. et al. The disease-ameliorating function of autoregulatory CD8 T cells is mediated by targeting of encephalitogenic CD4 T cells in experimental autoimmune encephalomyelitis. J Immunol 191, 117, doi: 10.4049/jimmunol (2013).
Ortega, S. B., Kashi, V. P., Cunnusamy, K., Franco, J. & Karandikar, N. J. Autoregulatory CD8 T cells depend on cognate antigen recognition and CD4/CD8 myelin determinants. Neurol Neuroimmunol Neuroinflamm 2, e170, doi: 10.1212/NXI.0000000000000170 (2015).
Teitelbaum, D., Meshorer, A., Hirshfeld, T., Arnon, R. & Sela, M. Suppression of experimental allergic encephalomyelitis by a synthetic polypeptide. Eur J Immunol 1, 242, doi: 10.1002/eji (1971).
Tennakoon, D. K. et al. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J Immunol 176, 7119, doi: 176/11/7119 (2006).
Tyler, A. F., Mendoza, J. P., Firan, M. & Karandikar, N. J. CD8 T Cells Are Required For Glatiramer Acetate Therapy in Autoimmune Demyelinating Disease. PLoS One 8, e66772, doi: 10.1371/journal.pone (2013).
Racke, M. K., Lovett-Racke, A. E. & Karandikar, N. J. The mechanism of action of glatiramer acetate treatment in multiple sclerosis. Neurology 74 Suppl 1, S25, doi: 10.1212/WNL (2010).
Bhowmick, S., Clark, R. B., Brocke, S. & Cone, R. E. Antigen-specific splenic CD4+ and CD8+ regulatory T cells generated via the eye, suppress experimental autoimmune encephalomyelitis either at the priming or at the effector phase. Int Immunol 23, 119, doi: 10.1093/intimm (2011).
Farooq, S. M., Elkhatib, W. F. & Ashour, H. M. The in vivo and in vitro induction of anterior chamber associated immune deviation to myelin antigens in C57BL/6 mice. Brain, behavior, and immunity 42, 118, doi: 10.1016/j.bbi (2014).
Cone, R. E., Chattopadhyay, S., Sharafieh, R., Lemire, Y. & O’Rourke, J. The suppression of hypersensitivity by ocular-induced CD8(+) T cells requires compatibility in the Qa-1 haplotype. Immunol Cell Biol 87, 241 (2009).
Bhattacharya, P. et al. Dual Role of GM-CSF as a Pro-Inflammatory and a Regulatory Cytokine: Implications for Immune Therapy. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research 35, 585, doi: 10.1089/jir (2015).
Bhattacharya, P., Gopisetty, A., Ganesh, B. B., Sheng, J. R. & Prabhakar, B. S. GM-CSF-induced, bone-marrow-derived dendritic cells can expand natural Tregs and induce adaptive Tregs by different mechanisms. J Leukoc Biol 89, 235, doi: 10.1189/jlb (2011).
Altman, J. D. et al. Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94 (1996).
Acknowledgements
We thank Dr. David Lo, a distinguished professor at the University of California at Riverside, for his critical reading, Penelope Garcia for her technical assistance and preparation of this manuscript. This work was supported by a Research Innovation Grant (RIG) from the Department of Medicine at Loma Linda University (681205-2967) and a pilot grant from National multiple Sclerosis Society (PP1685) to X.T.
Author information
Authors and Affiliations
Contributions
X.W., J.Z., C.-H.L. and Y.X. performed the experiments. X.T., D.J.B., D.M.W., X.Q. and M.H.W. designed the experiment. X.T. conceived and directed the study. All authors reviewed the manuscript.
Ethics declarations
Competing interests
XT and DJB are listed as inventors in a pending patent application related to this study.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, X., Zhang, J., Baylink, D. et al. Targeting Non-classical Myelin Epitopes to Treat Experimental Autoimmune Encephalomyelitis. Sci Rep 6, 36064 (2016). https://doi.org/10.1038/srep36064
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36064
This article is cited by
-
Innate, innate-like and adaptive lymphocytes in the pathogenesis of MS and EAE
Cellular & Molecular Immunology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
