
Abstract
As a potential alternative to antibiotics, phages can be used to treat multi-drug resistant bacteria. As such, the biological characteristics of phages should be investigated to utilize them as effective antimicrobial agents. In this study, phage PaoP5, a lytic virus that infects Pseudomonas aeruginosa PAO1, was isolated and genomically characterized. PaoP5 comprises an icosahedral head with an apex diameter of 69 nm and a contractile tail with a length of 120 nm. The PaoP5 genome is a linear dsDNA molecule containing 93,464 base pairs (bp) with 49.51% G + C content of 11 tRNA genes and a 1,200 bp terminal redundancy. A total of 176 protein-coding genes were predicted in the PaoP5 genome. Nine PaoP5 structural proteins were identified. Three hypothetical proteins were determined as structural. Comparative genomic analyses revealed that seven new Pseudomonas phages, namely, PaoP5, K8, C11, vB_PaeM_C2-10_Ab02, vB_PaeM_C2-10_Ab08, vB_PaeM_C2-10_Ab10 and vB_PaeM_C2-10_Ab15, were similar to PAK_P1-like viruses. Phylogenetic and pan-genome analyses suggested that the new phages should be assigned to PAK_P1-like viruses, which possess approximately 100 core genes and 150 accessory genes. This work presents a detailed and comparative analysis of PaoP5 to enhance our understanding of phage biology.
Similar content being viewed by others

Introduction
Bacteriophages or phages are abundant viruses that infect bacteria. The number of phages is approximately 10-fold higher than that of bacteria1. Since their discovery in 1915, phages have influenced basic and applied biology2. Since 1959, nearly 6,300 different phages have been examined through electron microscopy, including 6,196 bacterial and 88 archaeal phages3. In October 2012, 759 phages, including 721 infecting bacteria and 38 infecting archaea, were completely sequenced4. In February 2016, the number of completely sequenced phages reached 2,012, including 1,935 infecting bacteria and 77 infecting archaea, as revealed by the data from the National Center for Biotechnology Information (Bethesda, MA, USA). This number is lower than that of completely sequenced bacteria, which reached 5,020 in February 2016, although the genome size of phages is less than that of bacteria. Novel phages should be characterized and genomically analyzed to obtain additional valuable data regarding phages and help enhance our understanding of the evolutionary relationships between phages and bacteria.
As a Gram-negative opportunistic pathogen, Pseudomonas aeruginosa is the leading cause of local and systemic nosocomial infections; in some cases, its infection is life threatening5. P. aeruginosa infections are difficult to treat with antibiotics because of its intrinsic multi-drug resistance6. Thus, the biological characteristics of P. aeruginosa phages should be investigated to eradicate this notorious pathogen7. P. aeruginosa phages are taxonomically diverse and genetically dissimilar; they have been widely considered for their application as therapeutic and typing agents8. As of February 2016, 141 complete genome sequences of Pseudomonas phages mostly infecting P. aeruginosa have become available in GenBank9. P. aeruginosa phages are classified into several distinct genera, namely, PAK_P1-like10, KPP10-like11 and PB1-like viruses12. With the rapid development of genome sequencing, numerous novel P. aeruginosa phages have been identified. However, most of these phages have remained unclassified. Therefore, novel P. aeruginosa phages should be characterized and classified to facilitate the understanding of the interactions between P. aeruginosa and its phages and to help develop new approaches that combat this versatile pathogen.
Results and Discussion
Biological features of PaoP5
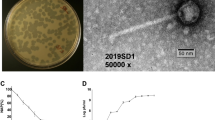
Phage PaoP5 was isolated from hospital sewage using P. aeruginosa PAO1 as host bacterium. PaoP5 was cultured overnight (~12 h) and formed large, clear plaques (~5 mm in diameter) on double agar plates. This finding suggested that PaoP5 is a lytic phage. Transmission electron microscopy analysis indicated that the head structure of PaoP5 is an icosahedron with an apex diameter of approximately 69 nm (Fig. 1). The non-contracted tail is about 120 nm in length. The contracted tail consists of a central tube, a 55 nm long contracted sheath and an 8 nm long neck. The morphological characteristics of phage PaoP5 suggest its membership under the Myoviridae family, members of which can affect many aspects of bacterial ecology and are efficient killers of bacteria, making them suitable for phage therapy13. Several attempts were made to explore the anti-bacterial potential of PAK_P1-like viruses14,15,16. New P. aeruginosa phages are continuously isolated and their capacity to target various clinical strains need to be tested in vitro and in animal models.

Electron micrograph of PaoP5 phage particles.
The sample was stained with phosphotungstate. The scale bar represents 100 nm. The black arrows indicate contracted tails.
Genomic characteristics of PaoP5
The length of the PaoP5 genome sequence is 93,464 bp, with an average G + C content of 49.51%, which is significantly less than that of its bacterial host (66.35%). The general features of the PaoP5 genome are listed in Table S1. Genome termini analysis revealed that PaoP5 holds a direct terminal repeat (DTR) with a length of approximately 1,200 bp (Fig. S1). The PaoP5 genome can be divided into six functional modules, of which two functionally unknown modules situate near the 5′ and 3′ ends of the PaoP5 genome, respectively and many small genes with unknown functions cluster in the two modules (Fig. S2). In addition, among the 176 predicted proteins of PaoP5, only 19.3% hold putative functions. Therefore, the vast number of phage genes with unknown functions should be explored extensively to better understand this interesting virus. The mosaic genome structure of PaoP5 suggests that its genome sequence may be evolved from combinations of modules from different species, similar to other tailed phage genomes17. The complete genome sequence and annotations of PaoP5 have been deposited in GenBank under the accession number KU297675.
Identification of phage PaoP5 structural proteins
To identify the structural proteins of PaoP5, sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) was used to separate and visualize each structural protein in the gel (Fig. 2A). Nine proteins with molecular weights ranging from 15 kDa to 76 kDa were determined. Then, each protein band was excised for high-performance liquid chromatography (HPLC)–mass spectrometry (MS), permitting the allocation of nine protein bands to nine corresponding PaoP5 genes. The detailed parameters and results of mass spectrometry are shown in Fig. 2B. The lowest sequence coverage was 15.63% for gp078, but the MS search score was not the lowest. Notably, the MS search score of gp055 was 0.00, indicating that the score should be further verified. Three hypothetical proteins, including gp055, gp064 and gp075 (Fig. 2B), were separated by SDS–PAGE, suggesting that these are actually structural proteins. Thus, we updated the corresponding GenBank records of gp055, gp064 and gp075 and conferred them with the function “structural protein” instead of “hypothetical protein.” As expected, the predominant band was predicted as the major capsid protein (gp057, ~39 kDa) of PaoP5. The structural proteins with molecular weight higher than 35 kDa, including tail fiber, baseplate and major capsid, are important for phage PaoP5 morphogenesis.

Identification of PaoP5 structural proteins.
(A) SDS–PAGE analysis. Proteins were visualized in a 15% (w/v) gel. (B) Detailed results of HPLC–MS analysis. aThe MW value was theoretically calculated. bThe MW value was experimentally estimated.
New members (including PaoP5) were assigned to PAK_P1-like viruses
BlastN analysis revealed that the genome sequences of phages PaP14,18,19,20, K8, C11, JG00421, vB_PaeM_C2-10_Ab114,22, vB_PaeM_C2-10_Ab0222, vB_PaeM_C2-10_Ab0822, vB_PaeM_C2-10_Ab1022, vB_PaeM_C2-10_Ab1522, PAK_P110,15,16, PAK_P210,16 and PAK_P410,16 share an identity of above 90% and query coverage also above 90% with the PaoP5 genome (Table S2 and Fig. S3). These similar phages were isolated in different areas around the world, spanning Asia, Europe and Africa (Fig. S4), suggesting the complex evolutionary relationships among these phages. The tBlastX analysis of the 13 phage genomes revealed that these phages show striking similarities at the protein level (Fig. S5).
As of February 3, 2016, 1,718 complete genomes of Caudovirales, including 463 Myoviridaes, 319 Podoviridaes, 913 Siphoviridaes and 23 unclassified Caudovirales were released in GenBank9. Among the 1,718 Caudovirales, 134 members infect Pseudomonas species (mostly P. aeruginosa). We conducted a phylogenetic analysis of 149 phages, including 134 members of Caudovirales that infect Pseudomonas species and 15 related members of Caudovirales that infect bacteria of other genera. The result indicated that the phages of the same genus are clustered into one clade, making the phylogenetic tree divide into several clades (Fig. 3). As expected, PaoP5 was clustered into PAK_P1-like viruses. In the year 2015, PAK_P1-like viruses was reported to have six members, including PaP1, JG004, vB_PaeM_C2-10_Ab1, PAK_P1, PAK_P2 and PAK_P410. Herein, we proposed that seven new members, including PaoP5, K8, C11, vB_PaeM_C2-10_Ab02, vB_PaeM_C2-10_Ab08, vB_PaeM_C2-10_Ab10 and vB_PaeM_C2-10_Ab15, should be assigned to PAK_P1-like viruses (Fig. 3). Although distributed in different clades, phages VCM, phiPsa374 and KPP10-like viruses are closely related to PAK_P1-like viruses (Fig. 3). Thus, all of these phages should be grouped into the subfamily of the Myoviridae named Felixounavirinae as proposed previously10.

Phylogenetic relationships of Caudovirales infecting Pseudomonas species.
Major capsid proteins and the neighbor-joining method were used to construct the phylogenetic tree. Different clades are marked with different colors. PAK_P1-like viruses are marked in red. The scale length of relative evolution distance is 0.2.
Given the 13 complete genomes of PAK_P1-like viruses, we performed a pan-genome analysis. Results showed that the pan-genome of PAK_P1-like viruses comprises approximately 100 core genes and 150 accessory genes (Fig. S6). Hence, new additional members of PAK_P1-like viruses are predicted to be characterized and sequenced in the near future. The core genome of this phage genus is expected to contain less than 100 genes, but infinitely close to a certain amount, which constitutes the minimal genome23 of these phages, thus providing useful clues for synthetic biology analysis24.
Materials and Methods
Bacterial strains and culture condition
P. aeruginosa PAO118,21 was used as the host bacterium of phage PaoP5. As for host spectrum assay of PaoP5, the tested 95 P. aeruginosa strains were isolated from the Department of Burn of the first affiliated hospital of the Third Military Medical University (Southwest Hospital, Chongqing, China) and cultivated in our laboratory. Bacteria were grown in Luria–Bertani (LB) liquid medium or plated onto solid LB medium containing 1.5% (w/v) agar and cultured at 37 °C with aeration.
Transmission electron microscopy (TEM)
Filtered phage lysates (~1011 PFU/mL) were placed on copper grids to allow adsorption for 10 min, then negatively stained with 2% phosphotungstic acid (PT-A, pH 4.5) for 1 min and subsequently air dried. Phage particles were observed using TECNAI 10 electron microscope (Philips, The Netherlands) at a voltage of 80 kV and with a magnification of 130,000. Images were acquired digitally with a camera (gatan Model 785) inside the microscope. Brightness and contrast were adjusted with Adobe Photoshop CS5.
SDS–PAGE and HPLC–MS of the PaoP5 structural proteins
Structural protein analysis was performed as described previously4. Briefly, the purified phage particles were heat-denaturized and loaded onto a 15% (w/v) polyacrylamide gel to visualize PaoP5 structural proteins. SDS–PAGE (12% [w/v] and 10% [w/v]) was also performed to better separate proteins with different molecular weights. Proteins were stained with Coomassie Brilliant Blue R250 dye and washed with methanol–acetic acid–H2O. Then, protein bands were excised from the gel for HPLC–MS analysis. The data from HPLC–MS analysis were processed by Agilent Spectrum Mill proteomics software to allocate each band to the corresponding gene.
Comparative genomic analysis
Thirteen complete phage genome sequences were subjected to BlastN comparisons by using blast 2.2.29+ (ftp://ftp.ncbi.nlm.nih.gov/blast/)25,26 and visualized by BRIG (http://brig.sourceforge.net/)27 with a 80% identity cut-off. The PaoP5 genome sequence was used as reference. Phage genome sequences were subjected to tBlastX analysis by using EasyFig (http://mjsull.github.io/Easyfig/)28 with a 33% identity cut-off. Major capsid protein sequences of phages (belonging to Caudovirales) infecting P. aeruginosa were downloaded from GenBank9. The multiple sequence alignments of major capsid protein sequences were conducted using ClustalW29 with default parameters and the phylogenetic tree was constructed and displayed by MEGA 6.06 (http://www.megasoftware.net/)30 with the neighbor-joining method31. We then constructed a Venn diagram using an online tool for calculating and drawing custom Venn diagrams (http://bioinformatics.psb.ugent.be/webtools/Venn/). Pan-genome analyses were performed using CoreGenes 3.5 Batch Submission Tool (http://binf.gmu.edu:8080/CoreGenes3.5/BatchCoreGenes.html)32 and Panseq (https://lfz.corefacility.ca/panseq/analyses#userPan)33 with default parameters and the results of CoreGenes and Panseq were combined to better present the pan-genome of PAK_P1-like viruses.
Additional InformationAccession code
Accession code: The complete genome sequence and annotations of PaoP5 have been deposited in GenBank under the accession number KU297675.
How to cite this article: Shen, M. et al. Characterization and Comparative Genomic Analyses of Pseudomonas aeruginosa Phage PaoP5: New Members Assigned to PAK_P1-like Viruses. Sci. Rep. 6, 34067; doi: 10.1038/srep34067 (2016).
References
Suttle, C. A. Marine viruses - major players in the global ecosystem. Nat Rev Microbiol 5, 801–812, 10.1038/nrmicro1750 (2007).
Salmond, G. P. C. & Fineran, P. C. A century of the phage: past, present and future. Nat Rev Microbiol 13, 777–786, 10.1038/nrmicr03564 (2015).
Ackermann, H. W. & Prangishvili, D. Prokaryote viruses studied by electron microscopy. Arch Virol 157, 1843–1849, 10.1007/s00705-012-1383-y (2012).
Lu, S. G. et al. Genomic and Proteomic Analyses of the Terminally Redundant Genome of the Pseudomonas aeruginosa Phage PaP1: Establishment of Genus PaP1-Like Phages. PLoS One 8, ARTN e62933 10.1371/journal.pone.0062933 (2013).
Hilker, R. et al. Interclonal gradient of virulence in the Pseudomonas aeruginosa pangenome from disease and environment. Environ Microbiol 17, 29–46, 10.1111/1462-2920.12606 (2015).
Breidenstein, E. B. M., de la Fuente-Nunez, C. & Hancock, R. E. W. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19, 419–426, 10.1016/j.tim.2011.04.005 (2011).
Vieira, A. et al. Phage therapy to control multidrug-resistant Pseudomonas aeruginosa skin infections: in vitro and ex vivo experiments. European journal of clinical microbiology & infectious diseases: official publication of the European Society of Clinical Microbiology 31, 3241–3249, 10.1007/s10096-012-1691-x (2012).
Ceyssens, P. J. & Lavigne, R. Bacteriophages of Pseudomonas. Future microbiology 5, 1041–1055, 10.2217/fmb.10.66 (2010).
Benson, D. A. et al. GenBank. Nucleic acids research 43, D30–D35, 10.1093/nar/gku1216 (2015).
Henry, M. et al. The search for therapeutic bacteriophages uncovers one new subfamily and two new genera of Pseudomonas-infecting Myoviridae. Plos One 10, e0117163, 10.1371/journal.pone.0117163 (2015).
Uchiyama, J. et al. Genetic characterization of Pseudomonas aeruginosa bacteriophage KPP10. Arch Virol 157, 733–738, 10.1007/s00705-011-1210-x (2012).
Ceyssens, P. J. et al. Comparative analysis of the widespread and conserved PB1-like viruses infecting Pseudomonas aeruginosa. Environ Microbiol 11, 2874–2883, 10.1111/j.1462-2920.2009.02030.x (2009).
Kropinski, A. M. Phage Therapy - Everything Old is New Again. The Canadian journal of infectious diseases & medical microbiolog = Journal canadien des maladies infectieuses et de la microbiologie medicale/AMMI Canada 17, 297–306 (2006).
Essoh, C. et al. The Susceptibility of Pseudomonas aeruginosa Strains from Cystic Fibrosis Patients to Bacteriophages. Plos One 8, ARTN e60575 10.1371/journal.pone.0060575 (2013).
Debarbieux, L. et al. Bacteriophages Can Treat and Prevent Pseudomonas aeruginosa Lung Infections. Journal of Infectious Diseases 201, 1096–1104, 10.1086/651135 (2010).
Henry, M., Lavigne, R. & Debarbieux, L. Predicting In Vivo Efficacy of Therapeutic Bacteriophages Used To Treat Pulmonary Infections. Antimicrobial agents and chemotherapy 57, 5961–5968, 10.1128/Aac.01596-13 (2013).
Hendrix, R. W., Smith, M. C., Burns, R. N., Ford, M. E. & Hatfull, G. F. Evolutionary relationships among diverse bacteriophages and prophages: all the world’s a phage. Proc. Natl. Acad. Sci. USA 96, 2192–2197 (1999).
Le, S. et al. Mapping the Tail Fiber as the Receptor Binding Protein Responsible for Differential Host Specificity of Pseudomonas aeruginosa Bacteriophages PaP1 and JG004. PLoS One 8, ARTN e68562 10.1371/journal.pone.0068562 (2013).
Le, S. et al. Chromosomal DNA deletion confers phage resistance to Pseudomonas aeruginosa. Scientific reports 4, Artn 4738 10.1038/Srep04738 (2014).
Lu, S. G. et al. Unlocking the mystery of the hard-to-sequence phage genome: PaP1 methylome and bacterial immunity. BMC genomics 15, Artn 80310.1186/1471-2164-15-803 (2014).
Garbe, J., Bunk, B., Rohde, M. & Schobert, M. Sequencing and Characterization of Pseudomonas aeruginosa phage JG004. Bmc Microbiol 11, Artn 10210.1186/1471-2180-11-102 (2011).
Essoh, C. et al. Investigation of a Large Collection of Pseudomonas aeruginosa Bacteriophages Collected from a Single Environmental Source in Abidjan, Cote d’Ivoire. Plos One 10, ARTN e013054810.1371/journal.pone.0130548 (2015).
Choe, D., Cho, S., Kim, S. C. & Cho, B. K. Minimal genome: Worthwhile or worthless efforts toward being smaller? Biotechnology journal 11, 199–211, 10.1002/biot.201400838 (2016).
Haynes, K. A. Synthetic biology: Building genetic containment. Nature chemical biology 12, 55–56, 10.1038/nchembio.2004 (2016).
Singh, H. & Raghava, G. P. S. BLAST-based structural annotation of protein residues using Protein Data Bank. Biol Direct 11, Artn 410.1186/S13062-016-0106-9 (2016).
Boratyn, G. M. et al. BLAST: a more efficient report with usability improvements. Nucleic acids research 41, W29–W33, 10.1093/nar/gkt282 (2013).
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC genomics 12, 402, 10.1186/1471-2164-12-402 (2011).
Sullivan, M. J., Petty, N. K. & Beatson, S. A. Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010, 10.1093/bioinformatics/btr039 (2011).
Hung, C. L., Lin, Y. S., Lin, C. Y., Chung, Y. C. & Chung, Y. F. CUDA ClustalW: An efficient parallel algorithm for progressive multiple sequence alignment on Multi-GPUs. Computational biology and chemistry 58, 62–68, 10.1016/j.compbiolchem.2015.05.004 (2015).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Molecular biology and evolution 30, 2725–2729, 10.1093/molbev/mst197 (2013).
Som, A. & Fuellen, G. The effect of heterotachy in multigene analysis using the neighbor joining method. Mol Phylogenet Evol 52, 846–851, 10.1016/j.ympev.2009.05.025 (2009).
Mahadevan, P., King, J. F. & Seto, D. CGUG: in silico proteome and genome parsing tool for the determination of “core” and unique genes in the analysis of genomes up to ca. 1.9 Mb. BMC research notes 2, 168, 10.1186/1756-0500-2-168 (2009).
Laing, C. et al. Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC bioinformatics 11, Artn 46110.1186/1471-2105-11-461 (2010).
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant 31400163 to S.Lu].
Author information
Authors and Affiliations
Contributions
The author(s) have made the following declarations about their contributions: S.Lu, F.H. and M.S. conceived and designed the experiments; M.S., S.Le, S.Lu, G.L., M.L., X.Z., Y.T. and W.S. performed the experiments; M.S., F.H., X.R., X.J., Y.Y., S.Li and J.W. analyzed the data; G.L., J.W., X.Z. and H.Z. contributed reagents/materials/analysis tools; M.S., F.H. and S.Lu wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shen, M., Le, S., Jin, X. et al. Characterization and Comparative Genomic Analyses of Pseudomonas aeruginosa Phage PaoP5: New Members Assigned to PAK_P1-like Viruses. Sci Rep 6, 34067 (2016). https://doi.org/10.1038/srep34067
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34067
This article is cited by
-
Isolation of a bacteriophage targeting Pseudomonas aeruginosa and exhibits a promising in vivo efficacy
AMB Express (2023)
-
Complete genome analysis of PaGz-1 and PaZq-1, two novel phages belonging to the genus Pakpunavirus
Archives of Virology (2020)
-
Characterization of the first double-stranded RNA bacteriophage infecting Pseudomonas aeruginosa
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
