
Abstract
This research is aimed at selecting specific aptamer of hepatitis B e antigen by SELEX and its applications. Hepatitis B e antigen (HBeAg) seroconversion is used as an indicator of virological response when treating patients suffering from chronic hepatitis B. HBeAg also indicates a high viremia and high infectivity in untreated patients. With HBeAg modified magnetic beads as targets, three groups of aptamers are successfully selected. These are the first reported DNA aptamers that can specifically bind to HBeAg. Based on the property that the conformation changes upon binding to its target, aptamer has emerged as ideal candidate in a variety of sensing applications. In this study, we present a simple strategy for aptamer-based fluorescence biosensors for the quantitative detection of HBeAg, in which a fluorescence labeled HBeAg aptamer serves as the molecular recognition element and a short DNA molecule that is complementary to the aptamer serves as the competitor. The LOD for HBeAg is 609 ng/mL. Later, the fluorescence system is deployed in HBeAg positive and negative blood serum (p < 0.05). The total detection assay could be completed in 2 min. These newly isolated aptamers could assist the diagnosis of chronic hepatitis B.
Similar content being viewed by others


Introduction
Hepatitis B is an infectious disease caused by the hepatitis B virus (HBV) that affects the liver. Recent estimates suggested that HBV infection caused 686 000 deaths in 20131,2. As 25% of people who acquire HBV as children will develop primary liver cancer or cirrhosis as adults, chronic hepatitis B virus infection (CHB) is a major public health issue worldwide3. However, current treatment options for CHB are ineffective4. Both pegylated interferon therapy and nucleos(t)ide analogues could not achieve the goal to cure CHB.
HBeAg is a viral protein of clinical importance, which is soluble and closely related to non-secretory capsid antigen hepatitis B core antigen (HBcAg). The sequences of those two proteins are largely overlapping. The different immunoreactivity between HBeAg and HBcAg is created by a different conformation of the B cell epitopes. HBeAg may also induce a specific non-responsiveness of helper T cells. HBeAg appears in the serum during the high replicative phase of HBV infection, which makes it a serum marker of active viral replication. It is also used to identify highly infectious mothers. Approximately 90% of infants of hepatitis B surface antigen (HBsAg) carrier mothers with positive HBeAg will become carriers if no immunoprophylaxis is given. Furthermore, HBeAg is accompanied by serum levels of HBV DNA that are 100,000 to 1 million IU per milliliter or higher5.
CHB can be broadly divided into two major forms: hepatitis B e antigen (HBeAg)-positive and HBeAg-negative. HBeAg-positive CHB is accompanied by high-level HBV replication, and spontaneous seroconversion from HBeAg-positive to antibody-positive infection coincides with a reduction in HBV replication and clinical improvement5. Patients with HBeAg-negative CHB harbor a naturally occurring mutant form of HBV that does not produce HBeAg because of a mutation in the precore or core promoter region of the HBV genome. These patients generally require prolonged treatment in contrast to those with HBeAg-positive CHB.
Aptamers are single-stranded nucleic acid ligands that are generated by an in vitro selection process called SELEX (systematic evolution of ligands by exponential enrichment)6,7. Aptamers can be considered as nucleic acid analogues of antibodies. They possess high affinities to target molecules, with dissociation constants in the nanomolar or picomolar range8. Currently, aptamers have been selected for a wide range of targets, including peptides, nucleotides, amino acids, antibiotics, proteins, vitamins, low-molecular weight organic or inorganic compounds, and even whole cells. Thus, aptamers are capable of becoming excellent chemical biosensors and may play an increasingly important role in public health, environmental monitoring, and food security9,10.
Several aptamers related to HBV have been previously selected. For example, Liu and colleagues reported the selection of RNA aptamers that can specifically bind to the HBsAg protein and HBsAg-positive hepatocytes11. Zhang and colleagues screened five different aptamers against HBcAg from a random ssDNA library, one of which could inhibit the assembly of the nucleocapsid and reduce extracellular HBV DNA, indicating its potential efficacy in suppressing HBV replication12. However, HBeAg-specific aptamers have not been reported.
Gold nanoparticles (AuNPs) possess strong distance-dependent optical properties and are considered important colorimetric materials13. AuNPs have been incorporated into novel biosensor strategies because of their optical properties, ease of use, and simple synthesis14. For example, Wang and colleagues reported a simple, sensitive, and inexpensive multiplex pathogen detection scheme based on novel gold nanorod bioprobes15. Sensors based on aptamer-linked AuNPs have also been demonstrated for detecting duplex DNA formation, protein-ligand interactions, and metal ion-ligand complexation16,17,18.
We also developed a rapid and sensitive method for the detection of HBeAg using aptamer-based fluorescence assay. Fluorescence molecules are attractive as a signaling moiety. Methods that employ fluorescent reporters have proven to be particularly useful in generation of aptamer-based biosensors. For example, fluorescence labeled aptamers have been reported for sensing a large amount of targets, such as thrombin19, mercury ions20, and adenosine21, etc.
The most typical detection method for HBeAg is ELISA (enzyme-linked immunosorbent assay). However, there are several disadvantages of ELISA, such as time-consuming and false positive/false negative deviation22. We selected novel aptamers that recognize and bind to HBeAg with appreciable affinity and selectivity. The most promising of the selected aptamers was incorporated with AuNPs and fluorescence, respectively, enabling the selective detection of HBeAg over other proteins.
Results
Test of binding affinity
ssDNA pools from each round were tested for their binding affinities to HBeAg (see Supplementary Fig. S1). The binding affinities of aptamers increased during later rounds of SELEX compared with those of the starting pool. However, binding affinities increased only slowly after nine rounds, indicating that affinity was nearing its maximum. Therefore, the SELEX process was terminated after the eleventh round.
Sequences and secondary structure of the selected aptamers
After eleven rounds, the selection process was completed, and the products were amplified, purified, and cloned. Twenty randomly picked clones were sequenced (Table 1). The results were divided into three groups. As can see in Fig. S2, each group of aptamers exhibited a stronger affinity for HBeAg immobilized on magnetic beads than that of aptamers from the starting pool. The affinity of aptamers from group 2 was found to be the strongest.
Secondary structures of the selected aptamers were analyzed by DNAMAN software (Fig. 1). Aptamers from group 2 shared the same secondary structure.

Predicted secondary structures of the core region of selected aptamers from groups 1, 2, and 3.
Detection of HBeAg using AuNP-based colorimetric aptasensor
As shown in Fig. S3, the 14-nm AuNPs used in our study are spherical and evenly distributed. Naked AuNPs aggregated to form bigger clusters in the presence of high concentration of NaCl. However, ssDNA-linked AuNPs did not aggregate. When 0.09 mM NaCl was added, the overall color of the solution changed from purple to red as the concentration of ssDNA increased (see Supplementary Fig. S4). Furthermore, when 4 μM ssDNA were added, the color of the solution was the same as that of the naked AuNPs when NaCl was not added. Therefore, we determined that the optimal concentration of added ssDNA was 4 μM.
The AuNP-based colorimetric aptasensor exhibited a maximum absorption peak at about 522 nm, as shown in Fig. 2. This indicates that the size of AuNP did not change after being coated with ssDNAs. Upon addition of HBeAg, the AuNPs combined with ssDNA to form a more stable aptamer-protein complex. This caused the release of ssDNA from the AuNPs. From these results, it could be concluded that aptamer-linked AuNPs aggregate to form bigger clusters in the presence of 0.09 mM NaCl and exhibit a new absorption peak with a maximum at higher wavelengths, whereas further increase in HBeAg concentration causes absorbance at 522 nm to decrease considerably. Importantly, this shift in the new peak becomes more obvious as AuNPs continue to aggregate into larger particles. Thus, in the presence of HBeAg, the aptasensor aggregated to form bigger clusters, and the overall solution color changed from red to dark purple. It should be noted that the addition of other proteins, such as trypsin and HBcAg did not cause the change in the color of the solution, indicating the high specificity of aptasensor for HBeAg (Fig. 3).

Detection of HBeAg by using an AuNP-based colorimetric aptasensor.

From left to right: aptamer-linked AuNPs + 0.09 M NaCl; aptamer-linked AuNPs + 50 nM parenzyme + 0.09 M NaCl; aptamer-linked AuNPs + 50 nM HBcAg + 0.09 M NaCl; aptamer-linked AuNPs + 50 nM HBeAg + 0.09 M NaCl.
Detection of HBeAg using fluorescence labeled aptamer
In this work, Dabcyl modified probe Q was used as fluorescence quencher. In the absence of HBeAg, a fluoresent dye (FAM) labeled HBeAg aptamer (named probe F) hybridized with probe Q to form a DNA duplex, leading to quenching. Once HBeAg was introduced, the FAM-aptamer was combind with HBeAg, which resulted in the FAM-aptamer being separated from probe Q and restoration of fluorescence. As shown in Table S1, the fluorescence intensity of aptamers decreased as the probe Q concentration was increased and eventually reached a saturation point after which any further increase in the probe Q concentration caused little decrease in the fluorescence intensity. Therefore, we determined the optimal concentration proportion of probe F and probe Q was 1: 3.
When HBeAg was added to the mixture of probe F and probe Q, the restoration of fluorescence was observed. As shown in Fig. 4, under the condition of 100 nM probe F, the linear relationship between the fluorescence intensity and the concentration of HBeAg was the best. Therefore, we determined the optimal concentration of probe F was 100 nM. To test the possibility of the fluorescence signal of the probe F for the quantitative detection of HBeAg, sufficient replicates were carried out. However, the addition of other proteins, such as trypsin, HBcAg and HBsAg did not cause the change in the fluorescence intensity, indicating the high specificity of the aptamer-based fluorescence biosensor for HBeAg (Fig. 5). Figure 6 showed that the fluorescence intensity increased as the concentration of HBeAg increased. The fluorescence signal had a good linear relationship with HBeAg concentration in the range of 42~420 nM, and the regression equation for this relationship was y = 2.6021 + 0.1274x (R = 0.9991). The limit of detection for HBeAg was as low as 26.5 nM.

The concentration of probe F was 50, 100 and 150 nM, respectively.

The concentration of proteins was 420 nM.

The linear relationship between the fluorescence intensity change and the HBeAg concentration.
Detection of HBeAg in blood serum using fluorescence labeled aptamer
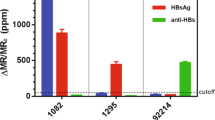
The fluorescence system was stored at 4 °C for two weeks and then deployed in diluted blood serum. Figure 7 showed that the fluorescence intensity increased sharply as the S/CO of HBeAg positive blood serum increased, and when the S/CO of blood serum reached 22.92, the rise trend began to slow down. As can been seen from the result, the fluorescence aptasensor was stable in two weeks. This proved the presented assay approach was simple and fast.

The mixture of probe F and probe Q was stored at 4 °C for two weeks or used immediately for detection. After gently shaking for 2 min, the fluorescence intensity was measured.
Discussion
We selected HBeAg-specific DNA aptamers using magnetic beads as an immobilization matrix for target molecules. Magnetic beads can provide a homogeneous surface with different functional groups for the immobilization of selected target molecules23,24. Additionally, unbound ssDNA was easily separated from bead-bound ssDNA through magnetic separation. This method offers the potential for parallel processing of multiple targets without the need for expensive robotics.
In this study, we have generated new HBeAg-specific aptamers and chosen the most promising aptamer for further development and application. Furthermore, an AuNP-based colorimetric aptasensor has been developed for colorimetric sensing of HBeAg. This method was based on HBeAg-aptamer interaction and distance-dependent optical properties of AuNPs. In the presence of HBeAg, the aptasensor aggregated to form bigger clusters, and the overall solution color changed from red to dark purple. Addition of other proteins did not cause the solution to change color, indicating that this aptasensor is highly specific for HBeAg. We also developed a rapid aptamer-based fluorescence biosensor to convert the aptamer-target recognition event into an fluorescence signal. In the presence of HBeAg, fluorescence recovery was observed. By taking advantage of affinity and specificity of aptamer, the biosensor showed excellent selectivity toward target. The biosensor also showed sensitivity and specificity to HBeAg in patient blood serum. As the fluorescence system was stable in two weeks or even more, the detection time could be shortened to 2 min.
Selection and application of HBeAg-specific aptamers described in this study is expected to promote the expoitation of aptamer-based detection for other proteins like HBsAg, EGFR (epithelial growth factor receptor) and so on. They can be designed as targeting ligands to differentiate diseased cells from healthy cells, thus enabling the selective delivery of therapeutic compounds to target cells25, such as aptamer-based DNA dendritic nanostructure as a multifunctional vehicle for targeted cancer cell imaging and drug delivery26. The emerging integration of nanotechnology and chemical biology is envisioned to produce further new clinical applications in the near future27.
Methods
Ethics statement
All experiments were carried out in accordance with relevant guidelines and regulations. The study was approved by the Research Ethics Committee of Southeast University, China. Because the tested samples were de-identified and would normally have been discarded. Informed consent of the study subjects was not required due to the nature of the samples.
Reagents and materials
The starting random ssDNA library and PCR amplification primers were synthesized by Shanghai Sangon Biotech Co. Ltd. (Shanghai, China). The ssDNA library contained a 40-base central random sequence flanked by primer sites on either side. The sequences of these designed oligonucleotides were as follows:
ssDNA library: 5′-GGGAATTCGAGCTCGGTACC-(40N)-CTGCAGGCATGCAAGCTTGG-3′.
Primer 1: 5′-GGGAATTCGAGCTCGGTACC-3′.
Primer 2: 5′-CCAAGCTTGCATGCCTGCAG-3′.
Primer 3: 5′-biotin-GGGAATTCGAGCTCGGTACC-3′.
Two DNA probe sequences were synthesized by Shanghai Sangon Biotech Co. Ltd. (Shanghai, China). The sequences were as follows:
Probe F: 5′-FAM-GGGGGGGCGAAGACCGGGACGGGAGGATTCTGTAGATTGGTTTT-3′. Probe Q: 5′-CTTCGCCCCCCC-Dabcyl-3′.
Bovine serum albumin (BSA), HEPES (free acid), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), and horseradish peroxidase (HRP)-conjugated streptavidin were purchased from Shanghai Sangon Biotech Co. Ltd. (Shanghai, China). Carboxylated magnetic beads were prepared by our laboratory. 2-N-morpholino ethanesulfonic acid (MES) and N-hydroxysuccinimide (NHS) were purchased from Life Science Products & Services. Recombinant HBeAg, HBsAg and HBcAg were purchased from Yikang Biological Technology Company (Zhengzhou, China). HBeAg positive and negative blood sera were kindly offered Gulou hospital (Nanjing, China).
HBeAg-coated magnetic beads were made by our laboratory.
Quick PCR Purification Kit was purchased from GenScript (Nanjing, China).
SELEX procedure and binding affinity assays
Specific procedures refer to previous article2,3 ssDNA pools with different concentrations (500 pmol for initial round; 100 pmol for 2nd–6th rounds; 50 pmol for 7th–11th rounds) were used for each selection step.
Preparation of AuNPs
A two-neck flask, magnetic stir bar, stopper, and condenser were soaked overnight in aqua regia. The glassware was rinsed with copious amounts of ddH2O twice. Five hundred microliters of 1% HAuCl4 solution were added into 49.5 ml of ddH2O so that the final HAuCl4 concentration was 0.01%. One neck of the flask was connected with the condenser and the other with the stopper. The flask was placed on a hot plate to reflux while stirring. When the solution began to boil, 1.5 mL of 1% sodium citrate was quickly added. The color changed from pale yellow to dark purple and then to deep red in 1 min. The system was allowed to reflux for another 20 min and then return to room temperature (25 °C) for 2 h. The prepared AuNPs were stored at 4 °C.
Detection of HBeAg using AuNPs-based colorimetric aptasensor
40 μL of the HBeAg-specific aptamer were added into 2.5 mL of AuNPs and incubated at room temperature for 20 min. The solution was divided into 4 groups. 1 μL each of ultrapure water, trypsin, HBcAg and HBeAg was added to 400 μL of nanoparticles in each group, respectively. After standing for 30 min, 0.09 mM NaCl was added. The absorbance of nanoparticles was measured by UV-visible spectroscopy (Shimadzu UV-1800).
Detection of HBeAg using fluorescence labeled aptamer
To determine the optimal concentration proportion of probe F and probe Q, two microliters each of ultrapure water and 5, 10, 15, 20 μM probe Q were added to a series of Eppendorf centrifuge tubes containing 200 μL of buffer A (50 mM NaCl, 7 mM MgCl2, 50 mM Tris-HCl) and 50 nM of probe F, respectively. After incubated at room temperature for 20 min, the fluorescence intensity was determined using a Synergy HT Multi-Mode Microplate Reader, with an excitation wavelength of 485 nm and an emission wavelength of 528 nm.
To demonstrate the detection capability of the fluorescence labeled aptamer, 1.2 μL of probe F (50 μM) was mixed with 3.6 μL of probe Q (50 μM) in 1.2 mL of buffer A. The solution was divided into 6 equal parts. Five microliters each of ultrapure water and different concentrations from 0.168 to 3.36 μM of HBeAg were added to each equal part, respectively. After standing for 30 min, the fluorescence intensity was measured.
To demonstrate the specificity of the fluorescence labeled aptamer, 5 μL of probe F (10 μM) was mixed with 15 μL of probe Q (10 μM) in 1 mL of buffer A. The solution was divided into 5 groups. 5 μL each of ultrapure water, 1.68 μM trypsin, HBsAg (hepatitis B surface antigen), HBcAg (hepatitis B core antigen) and HBeAg were added to each equal part, respectively. After standing for 30 min, the fluorescence intensities were measured.
To determine the optimal concentration of probe F, five microliters each of 50, 100 and 150 μM probe F were added to a series of Eppendorf centrifuge tubes containing 1 mL of buffer A. And after 20 min incubating with probe Q, the solution in each tube was divided into 5 partes aequales. Five microliters each of ultrapure water and different concentrations from 0.168 to 3.36 μM of HBeAg were added to each equal part. After standing for 30 min, the fluorescence intensities were measured.
Detection of HBeAg in blood serum using fluorescence labeled aptamer
460.8 μL of probe F (10 μM) was mixed with 1382.4 μL of probe Q (10 μM) in 7.2 mL of buffer A. The solution was then divided into 72 equal parts. 40 μL each of HBeAg positive blood serum were added to each equal part, respectively. After gently shaking for 2 min, the fluorescence intensity was measured. To test the stability of the fluorescence based aptasensor, the mixture of probe F and probe Q was stored at 4 °C for two weeks and then used for detection.
Additional Information
How to cite this article: Huang, R. et al. Fluorescence based Aptasensors for the determination of hepatitis B virus e antigen. Sci. Rep. 6, 31103; doi: 10.1038/srep31103 (2016).
References
Lozano, R. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128 (2013).
MacLachlan, J. H., Locarnini, S. & Cowie, B. C. Estimating the global prevalence of hepatitis B. Lancet 386, 1515–1517 (2015).
Schweitzer, A., Horn, J., Mikolajczyk, R. T., Krause, G. & Ott, J. J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386, 1546–1555 (2015).
Lampertico, P., Maini, M. & Papatheodoridis, G. Optimal management of hepatitis B virus infection-EASL Special Conference. J Hepatol 63, 1238–1253 (2015).
Dienstag, J. L. & Hepatitis, B. virus infection. New Engl J Med 359, 1486–1500 (2008).
Clark, S. L. & Remcho, V. T. Aptamers as analytical reagents. Electrophoresis 23, 1335–1340 (2002).
Tuerk, C. & Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510 (1990).
Ellington, A. D. & Szostak, J. W. In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 (1990).
Sefah, K. et al. Nucleic acid aptamers for biosensors and bio-analytical applications. Analyst 134, 1765–1775 (2009).
Fan, R. J. et al. Reliable screening of pesticide residues in maternal and umbilical cord sera by gas chromatography-quadrupole time of flight mass spectrometry. Sci China Chem 57, 669–677 (2014).
Zeng, Z. et al. Specific and Sensitive Tumor Imaging Using Biostable Oligonucleotide Aptamer Probes. Theranostics 4, 495–952 (2014).
Liu, J. et al. Development of HBsAg-binding aptamers that bind HepG2.2.15 cells via HBV surface antigen. Virol Sin 25, 27–35 (2010).
Zhang, Z. et al. An aptamer targets HBV core protein and suppresses HBV replication in HepG2.2.15 cells. Int J Mol Med 34, 1423–1429 (2014).
Pan, Y. et al. Size-Dependent Cytotoxicity of Gold Nanoparticles. Small 3, 1941–1949 (2007).
Sattarahmady, N., Tondro, G. H., Gholchin, M. & Heli, H. Gold nanoparticles biosensor of Brucella spp. genomic DNA: Visual and spectrophotometric detections. Biochem Eng J. 97, 1–7 (2015).
Wang, C. & Irudayaraj, J. Gold Nanorod Probes for the Detection of Multiple Pathogens. Small 4, 2204–2208 (2008).
Jiang, Z. et al. A highly selective nanogold-aptamer catalytic resonance scattering spectral assay for trace Hg2+ using HAuCl4-ascorbic acid as indicator reaction. Talanta 80, 1287–1291 (2010).
Emir, Y. et al. Enrichment and Detection of Rare Proteins with Aptamer-Conjugated Gold Nanorods. Anal Chem 84, 6608–6615 (2012).
Wei, H., Li, B., Li, J., Wang, E. & Dong, S. Simple and sensitive aptamer-based colorimetric sensing of protein using unmodified gold nanoparticle probes. Chem Commun 36, 3735–3737 (2007).
Yu, J., Yang, L., Liang, X., Dong, T. & Liu, H. Bare magnetic nanoparticles as fluorescence quenchers for detection of thrombin. Analyst 140, 4114–4120 (2015).
Jiang, Y., Tian, J., Hu, K., Zhao, Y. & Zhao, S. Sensitive aptamer-based fluorescence polarization assay for mercury(II) ions and cysteine using silver nanoparticles as a signal amplifier. Microchimi Acta 181, 1423–1430 (2014).
Wang, S. H. & Si, S. H. Direct fluorescence polarization aptamer-based assay for the determination of adenosine. Anal Methods 5, 813–1072 (2013).
Menghua, L. Comparison of two methods for the detection of HBeAg in the serum of 386 cases. China Medical Herald 6, 75–75 (2009).
Wang, C. L., Yang, G., Luo, Z. F. & Ding, H. M. In vitroselection of high-affinity DNA aptamers for streptavidin. Acta Biochim Biophys Sin 41, 335–340 (2009).
Xi, Z. et al. Selection of HBsAg-Specific DNA Aptamers Based on Carboxylated Magnetic Nanoparticles and Their Application in the Rapid and Simple Detection of Hepatitis B Virus Infection. ACS Appl Mater Inter 7, 11215–11223 (2015).
Yang, L. et al. Photothermal Therapeutic Response of Cancer Cells to Aptamer-Gold Nanoparticle-Hybridized Graphene Oxide under NIR Illumination. ACS Appl Mater Inter 7, 5097–5106 (2015).
Zhang, H. et al. A controllable aptamer-based self-assembled DNA dendrimer for high affinity targeting, bioimaging and drug delivery. Sci Rep 5 (2015).
Matsunaga, K. I. et al. Architecture of high-affinity unnatural-base DNA aptamers toward pharmaceutical applications. Sci. Rep 5 (2015).
Acknowledgements
This research was financially supported by the National Key Program for Developing Basic Research (2014CB744501), the Chinese National Key Project of Science and Technology (2013ZX10004103-002), the NSFC (61271056, 61471168, 61201100 and 61527806), and the Economical Forest Cultivation and Utilization of 2011 Collaborative Innovation Center in Hunan Province [(2013) 448].
Author information
Authors and Affiliations
Contributions
R.H. and Z.X. wrote the main manuscript text. Y.D. and N.H. prepared Figure 1, Table 1 and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Huang, R., Xi, Z., Deng, Y. et al. Fluorescence based Aptasensors for the determination of hepatitis B virus e antigen. Sci Rep 6, 31103 (2016). https://doi.org/10.1038/srep31103
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31103
This article is cited by
-
Multiple virus sorting based on aptamer-modified microspheres in a TSAW device
Microsystems & Nanoengineering (2023)
-
Screening of hepatitis B virus DNA in the serum sample by a new sensitive electrochemical genosensor-based Pd-Al LDH substrate
Journal of Solid State Electrochemistry (2022)
-
Recent developments in affinity-based selection of aptamers for binding disease-related protein targets
Chemical Papers (2019)
-
Aptamer selection and applications for breast cancer diagnostics and therapy
Journal of Nanobiotechnology (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
