
Abstract
Traditional cheeses harbour complex microbial consortia that play an important role in shaping typical sensorial properties. However, the microbial metabolism is considered difficult to control. Microbial community succession and the related gene expression were analysed during ripening of a traditional Italian cheese, identifying parameters that could be modified to accelerate ripening. Afterwards, we modulated ripening conditions and observed consistent changes in microbial community structure and function. We provide concrete evidence of the essential contribution of non-starter lactic acid bacteria in ripening-related activities. An increase in the ripening temperature promoted the expression of genes related to proteolysis, lipolysis and amino acid/lipid catabolism and significantly increases the cheese maturation rate. Moreover, temperature-promoted microbial metabolisms were consistent with the metabolomic profiles of proteins and volatile organic compounds in the cheese. The results clearly indicate how processing-driven microbiome responses can be modulated in order to optimize production efficiency and product quality.
Similar content being viewed by others

Introduction
Cheese is a biologically and biochemically dynamic matrix, where the microbiota structure and activity are influenced by manufacture practices and environmental conditions. Traditional cheeses either employ naturally selected starter cultures with extensive genetic diversity, or go without any starter1,2. Therefore, traditional cheeses harbour a rich and complex microbiota, arising from the use of undefined natural whey cultures (NWCs) used according to a back-slopping procedure, but also from raw milk and from the dairy environment3, whose role during manufacture and ripening is recognized but often under-explored. Cheese manufacture is characterized by a succession of different lactic acid bacteria (LAB) species. Curd acidification is mainly driven by thermophilic LAB, mostly Streptococcus thermophilus, Lactobacillus delbrueckii and Lb. helveticus2,4,5, while mesophilic non-starter LAB (NSLAB) take over during the ripening and can be involved in the development of cheese flavour and texture6,7,8,9,10,11. The microbiota plays an important role in shaping the typical sensorial properties of cheese12,13. Nevertheless, these complex microbial consortia are often difficult to control and their dynamics and evolution during cheese making and ripening cannot be easily predicted. Understanding microbial behaviour during cheese ripening and how the technological parameters applied can influence the cheese making dynamics and the quality of the final product, are pivotal steps in order to ensure safety and quality3,14. Moreover, accelerating the ripening kinetics through the modulation of microbial activities can be extremely important for traditional, small-scale and more industrialized, dairies. With this control they can reduce operating costs, increase the turnover of ripening chambers and optimize the process efficiency having cheese quality as priority. Studying the dynamic microbial ecology and metabolism during cheese making using multi-omic approaches may help to improve efficiency14,15. Metatranscriptomics has been recently used to study inoculated strain cultures in model cheeses16,17, yet the transcript dynamics of complex communities remains unexplored. On the other hand, fermented food microbial communities are not so complex as those of other natural environments and may be a replicable and tractable system to study the dynamics of microbial assembly and how they may be manipulated by abiotic factors18.
Longitudinal characterisation of the microbial community structure and gene expression was performed during ripening of the traditional Italian Caciocavallo Silano cheese. This is a typical medium-ripened pasta-filata cheese produced with the addition of NWC as natural starter. Initial investigation, reported here, identified parameters that could be modified to accelerate ripening. Therefore, we set up a second experiment, in which the ripening conditions were modulated and consistent changes in microbial community structure and function were observed. We reveal how increasing ripening temperature affects microbial metabolism, which significantly increases the cheese maturation rate.
Results
Cheese ripening was explored in two separate experiments. Firstly, microbial community structure (16S rRNA amplicon) analysis and a shallow exploration of gene expression (metatranscriptomics) were performed on raw and thermized cow milk, natural whey culture, curd after 5 hours of fermentation and before stretching, cheeses after brining and during the ripening at 0, 10, 20, 30 and 60 days. In this way, we identified which bacterial activities/pathways were highly expressed during production and ripening of a pasta-filata cheese, which provided the parameters necessary to understand how to accelerate ripening.
Then, we set up a second experiment, characterising the microbiome across 30 days, for cheeses ripened in three different conditions: (A) standard conditions in the dairy (16 °C and 75% RH), (B) increasing the temperature to 20 °C and (C) decreasing the RH to 65%.
We obtained a total of 3.3 Gbp in the first experiment and 19.8 Gbp in the second experiment. In the first experiment, an average of 876,295 reads/sample (±409,657) were obtained and an average of 22% of reads uniquely mapped to the reference genomes (ranging from 12 to 27.8%).
In the second experiment we obtained an average of 1,180,7044 reads/sample (±5.9 millions) and an average of 35.5% of reads uniquely mapped to the reference genomes (ranging from 23.9 to 58.6%). The normalized abundance (reads per million) of the genes identified for each species and the OTU relative abundance (%) are reported as Supplementary Table S1 and S2.
Non-Starter Lactic Acid Bacteria (NSLAB) increase in relative abundance both during cheese maturation and with ripening temperature
Cheese ripening was driven by few non-starter lactobacilli, e.g. Lb. casei and Lb. buchneri, groups whose abundance was greater in the cheese core compared to the crust. This result was consistent in both the experiments carried out in this study. Firmicutes were the phylum that most significantly differentiated the core and crust microbiota (Supplementary Fig. 1). Microbial loads on MRS agar incubated at 30 °C increased during the ripening, with greater values in the cheese core, while thermophilic lactobacilli and streptococci progressively decreased (Supplementary Table 3). When we compared three different ripening conditions (second experiment), NSLAB at 10 days of ripening were significantly more abundant when relative humidity was reduced (C–17.2%) and when ripening temperature was increased (B–15.3%) compared to the control conditions (A–8.3%) (P < 0.05). However, in the reduced relative humidity condition (C), NSLAB abundance rapidly decreased after 10 days of ripening. Interestingly, Lb. fermentum, which was not detected in the control condition, appeared in the increased temperature condition (B) after 10 days and continued to increase in abundance throughout ripening.
Ripening-associated microbial metabolism is greater in the cheese core, increases with temperature and is consistent with aroma profiles
The results from the first experiment showed that curd and cheese after moulding and brining were characterized by the prevalence of genes involved in carbohydrate metabolism. During the ripening, carbohydrate metabolism (pentose-phosphate pathway, glycolysis) and cellular division (genetic information processing and core cellular processes) were enriched on cheese crusts, while cheese cores were characterized by significantly elevated amino acid and lipid metabolism (Fig. 1). In addition, samples ripened at higher temperature (B) during the second experiment showed an increase in the expression of pathways related to amino acid and lipid metabolism (Fig. 2a) and associated genes (Fig. 2b) compared to the control (A) at the same ripening times. Genes related to the Leloir pathway of galactose degradation were over-expressed compared to the tagatose-6-phosphate pathway. Enzymes responsible for lactose break-down (lacZ, EC 3.2.1.23 and lacG, EC 3.2.1.85), Leloir and tagatose pathways showed maximum expression in the early stages of manufacture and decreased during ripening (Supplementary Fig. 2). Enzymes leading to acetoin and diacetyl production were up-regulated on the crust and had significantly greater expression at the higher temperature (B compared to A; Supplementary Fig. 3). Moreover, a number of peptidases, amino acid and peptide permeases, lipases and genes involved in amino acid catabolism and β-oxidation of fatty acids were over-expressed in the core of cheeses ripened at higher temperature (Fig. 2b and Supplementary Table 4). Finally, higher temperature boosted the expression of genes involved in fatty acids biosynthesis from pyruvate (Fig. 3). The DESeq analysis identified 651 genes differentially expressed between the cheese cores ripened in condition A and B (P value < 0.05) regardless of the ripening time (Supplementary Table 4). Among them, the proteases, peptidases, dipeptide transporters, amino acid permeases, amino acid catabolism genes, fatty acid β-oxidation and biosynthesis were over-expressed in the core of samples B compared to A (Supplementary Table 4).

Metatranscriptome drives cheese core and crust separation.
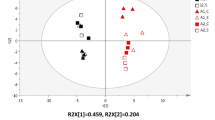
PCA based on the abundance of the KEGG annotations at level 2 of hierarchy of the samples of cheese core and crust from the first experiment carried out in this study. The first component (horizontal) accounts for the 65.7% of the variance and the second component (vertical) accounts for the 12.2%. CO, samples of cheese core; CR, samples of cheese crust.

Higher temperature promotes ripening-related gene expression.
Average-linkage clustering of samples from the second experiment based on the Euclidean distance of the proportion of the KEGG pathways (upper) or genes (lower) belonging to carbohydrates (violet), amino acids (orange) and lipids (cyan) metabolisms. Column bar is color-coded as follows: red, cheese at t0; blue, cheese ripened in standard conditions, green, cheese ripened at higher temperature. The color scale represents the scaled abundance of each variable, denoted as Z-score, with red indicating high abundance and blue indicating low abundance. In the sample IDs, the first part indicates the time of ripening (from t0, cheese after brining and drying, to 30 days of ripening); CO, core or CR, crust; A, ripening at standard conditions; B, higher temperature.

Pathways leading to fatty acids production from amino acids.
Biosynthesis of fatty acids from pyruvate (A) and from branched-chain amino acids catabolism (B). Only KEGG genes identified in the samples analyzed are reported and their abundance is shown in the heatplot (C). Average-linkage clustering is based on the Spearman distance of the proportion of the KEGG genes. Only samples from the second experiment are shown. Column bar is color-coded as follows: red, cheese at t0; blue, cheese ripened in standard conditions; green, cheese ripened at higher temperature. The color scale represents the scaled abundance of each variable, denoted as Z-score, with red indicating high abundance and blue indicating low abundance. In the sample IDs, the first part indicates the time of ripening (from t0, cheese after brining and drying, to 30 days of ripening); CO, core or CR, crust; A, ripening at standard conditions; B, higher temperature. *Malonyl-CoA can be substituted by propanoyl-CoA, leading to odd-numbered chain fatty acids. **For each cycle, 2 carbon atoms are added.
Amino acid metabolism expression was predominantly associated with the Firmicutes, suggesting a clear effect of temperature on the metabolism of this phylum (Fig. 4). Specifically, the amino acid metabolism expression of Lb. casei appeared to be significantly increased under the elevated temperature. However, other NSLAB taxa (such as Lb. buchneri, Lb. plantarum, Lb. gasseri, Lb. fermentum, Lb. rhamnosus, Leuconostoc kimchii, Leuc. mesenteroides, Leuc. citreum, Pediococcus pentosaceus) also showed increased amino acid metabolism activity at the higher temperature. A network showing significant (FDR < 0.05) Spearman’s correlations between KEGG genes, OTUs and metabolome clearly suggests that the abundance of NSLAB was positively correlated to genes involved in amino acid and fatty acid metabolism, as well as the abundance of volatile compounds, lipolysis and proteolysis indices (Fig. 5). Moreover, ripening-associated activities were inversely related to the abundance of thermophilic LAB from the natural starter (Fig. 5).

Higher temperature boosts NSLAB amino acid metabolism.
Taxonomic assignment of the genes belonging to KEGG amino acid metabolism in the samples of cheese core from the second experiment, at 30 days of ripening. Only species belonging to Firmicutes are reported. A, ripening at standard conditions; B, higher temperature.

NSLAB abundance, ripening-related gene expression and metabolome are strongly linked.
Network showing significant (FDR < 0.1) Spearman’s correlations between KEGG genes belonging to amino acid and lipid metabolism, VOCs, lipolysis and proteolysis indices and OTUs belonging to Firmicutes identified through 16S rRNA sequencing. Node size was made proportional to the number of significant correlations. Edge color indicates negative (blue) or positive (grey) correlations. Node color was assigned as follows: green, KEGG genes related to aminoacid metabolism; red, KEGG genes related to lipid metabolism; yellow, VOCs; magenta, chemical indices; cyan, Firmicutes OTUs.
The higher ripening temperature also promoted the development of flavour compounds, both in the cheese core and on the crust (Table 1). The metabolomic profiles were highly consistent with the expression profiles from the metatranscriptomic analysis. Volatile short-chain fatty acids (butanoic, pentanoic, hexanoic, heptanoic, octanoic and decanoic acids) were the most abundant volatiles, together with 2-methyl ketones and alcohol compounds, such as hexanol, 2-heptanol, 1-butanol and 3-methyl butanol. Methyl ketones, secondary alcohols and free fatty acids increased in abundance during ripening and were elevated at the higher temperature, which was also observed for 3-methyl butanol (P < 0.05). Free fatty acids may arise from direct lipolysis of triacylglycerols, or they may be synthesized ex-novo from pyruvate evolved from amino acid catabolism (Fig. 3). Lipolysis and proteolysis indices were greater in the core and increased in cheese ripened at higher temperature (Table 1). Accordingly, LC/MS analysis of the pH 4.6 insoluble nitrogen fraction showed an extensive degradation of either αs1- and β-casein in the cheese ripened at higher temperature, while the pH 4.6-soluble fraction showed a greater amount of soluble peptides derived from the two proteins (Supplementary Fig. 4).
Discussion
The cheese ripening process is very complex and involves microbiological and biochemical changes leading to the development of a specific flavour and texture. The cheese microbiome is shaped by the abiotic factors encountered during ripening (pH, aw, NaCl concentration, O2 availability). Therefore, different dynamics may occur in the cheese core and crust, due to the different environmental conditions3,19. The importance of fungi- and bacteria-associated metabolic activities in surface-ripened cheeses was recently highlighted16,17,20, while an increase in NSLAB in ripened cheeses was previously observed7,10. NSLAB are known as proteolytic21,22,23 and the adjunct of selected cultures was shown to increase proteolysis levels8,24,25, suggesting they may play a key role during cheese ripening. We provide concrete evidence of their essential contribution, showing that a different structure and functionality of the microbiome evolves in cheese core and crust and that the ripening proceeds with a gradient starting from the core towards the crust, driven by NSLAB succession. On the cheese surface, where lower aw and higher O2 concentrations make the environment adverse, NSLAB development is delayed and the cellular metabolism is directed towards multiplication and energy production from carbohydrates. Consistently, lactose break-down and galactose metabolism were retarded on the cheese crust and their levels kept constantly higher on the crust compared to the core. Moreover, microbial activities are strongly influenced by the conditions applied during ripening. We cannot exclusively link the higher proteolysis levels to bacterial activity and endogenous enzymes may be still active in cheese without a curd-cooking step. However, a clear bacterial contribution to ripening activities is supported by the consistency between metatranscriptome and metabolome data and by the significant degree of correlation between the occurrence of NSLAB, expression profiles and metabolic patterns (Fig. 5). Particularly, bacterial gene expression of ripening-associated activities is significantly linked to metabolite profiles and proteolysis and lipolysis indices.
The increase in proteolysis, lipolysis and amino acids/fatty acids catabolism boosted by the higher temperature definitely promoted the production of volatile compounds. The observed free fatty acids may arise from direct lipolysis of triacylglycerols, as well as from ex novo biosynthesis from pyruvate produced by amino acid catabolism26. Moreover, 3-methyl butanol is produced by leucine degradation and methyl ketones and secondary alcohols come from fatty acids catabolism27,28.
Recently, the importance of exploring microbial assembly mechanisms in fermented foods (MCoFFs) was highlighted18. Fermented foods give tantalizing opportunities to study microbial metabolic patterns and ecological dynamics, identifying the major drivers and testing hypotheses that can be easily extrapolated to more complex environments18. We give a clear example of how they might be used to understand the effect of abiotic factors on microbial community assembly and metabolism. This supports the potential for modelling microbial community dynamics in more complex environments18.
The increase in ripening temperature promotes the expression of genes related to proteolysis and amino acid/lipid transport and catabolism, leading to a higher concentration of free amino acids and fatty acids and boosting the production of VOC typically found in Caciocavallo cheese volatilome7, possibly accelerating the ripening and enhancing the potential flavour impact of the cheese. Indeed, cheeses ripened at higher temperature for only 20 days had very similar transcriptomic profiles as those ripened for longer at standard temperature, indicating that the ripening-related activity is boosted by the change in ripening conditions. Finally, taxonomic assignment of genes related to KEGG amino acid metabolism pathways confirmed a shift in the amino acid metabolism due to the higher temperature and emphasized the role of NSLAB as fundamental players in cheese maturation.
The results clearly indicate how changes in ripening conditions are important to manipulate cheese microbiome and its activities and that the response of the microbiome can be promptly used to improve processing conditions and product quality.
Methods
Cheese manufacture and sampling
The cheese manufacture and ripening was carried out in a dairy located in Southern Italy and producing Caciocavallo Silano cheese granted the Protected Designation of Origin (PDO, CE Reg. 1263/96). In a first experiment, raw and thermized cow milk, NWC, curd after 5 h of fermentation and before stretching (when pH reached ca. 5.25), cheeses after molding, after brining and during the ripening (at 10, 20, 30 and 60 days) were collected and analyzed through 16S rRNA sequencing and RNA-seq (Supplementary Table 5). In a second experiment, in order to evaluate the effect of ripening parameters on the microbiome, we decided to ripe the cheeses from the same manufacturing in three different conditions: a control ripening (A) by using the standard conditions in the dairy (16 °C and 75% RH, condition A), increasing the temperature to 20 °C (B) or decreasing the RH to 65% (C). In this second experiment, the ripened cheeses were collected up to 30 days. Sample IDs reported in Supplementary Table 5 and throughout the text and the figures were assigned as follows: the first part of the ID indicates the time of ripening (from t0, cheese after brining and drying, to 60 days of ripening); CO, core or CR, crust; A, ripening in standard conditions, B, ripening at higher temperature, C, ripening at lower RH.
Water activity (aw) was measured by using a HygroPalm23-AW instrument (Rotronic AG, Basserdorf).
Samples from the core (the inner part, collected in the middle of the cheese) and crust (the outer part, after peeling the external layer) were cut (in slices of ~0.5 cm thick) in sterile conditions and analyzed separately. Different aliquots from each sample were collected in sterile plastic bags for the different analyses and transported to the laboratory under refrigerated conditions (4 °C). RNAlater (Ambion, Foster City, California) was added in 1:6 ratio to the aliquot to be used for RNA extraction before transport. Samples were stored at −80 °C prior to analyses, except for microbial counts, that were carried out within 2 h from the sampling, as follows.
Twenty-five grams (solid) or 25 ml (liquid) of sample at each time of sampling and under each ripening condition were homogenized in 225 ml of quarter-strength Ringer’s solution (Oxoid, Milan, Italy) for 2 min in a stomacher (LAB Blender 400) by using Sto-Circul-Bag stomacher bags (both from PBI, Milan, Italy) at room temperature. Decimal dilutions in quarter-strength Ringer’s solution were prepared and 1 ml aliquots of the appropriate dilutions were inoculated in triplicate in M17 or MRS agar (Oxoid, Milan, Italy) and incubated aerobically and anaerobically, respectively, at 30 or 42 °C for 48 h to obtain the viable counts of mesophilic and thermophilic streptococci or lactobacilli. Results were calculated as the means of log colony forming units (CFU)/g for three independent determinations.
Samples preparation and RNA extraction
Samples stored in RNA later were homogenized and 10 ml of the mixture was centrifuged for 10 min at 4 °C (12,000 × g). Two biological replicates (two different cheeses at each sampling point and for each condition) were subjected to RNA extraction and total RNA was pooled before further processing. The pellet was washed twice in PBS (Phosphate-Buffered Saline, pH 7.4) and RNA extraction was carried out in duplicate by using the PowerMicrobiome RNA Isolation kit (MoBio, Carlsbad, California). DNA was removed by a treatment with TURBO-DNase (Ambion, Foster City, California) for 3 h at 37 °C. The absence of DNA was checked by PCR and the treatment repeated if necessary. The quality of the RNA was checked by agarose gel electrophoresis and by the 2100 Bioanalyzer (Agilent Technologies, Palo Alto, California). The Qubit and the Qubit RNA Assay kit (Life Technologies, Carlsbad, California) were used for quantification.
16S rRNA library preparation and amplicon sequencing
Complimentary DNA (cDNA) was synthetized by using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, California), starting from 200 ng of total RNA. The microbial diversity was studied by pyrosequencing of the amplified V1–V3 region of the 16S rRNA as reported elsewhere29. PCR conditions, library preparation and sequencing were carried out as recently described30.
Ribosomal RNA depletion, library preparation and shotgun metatranscriptome sequencing
Metatranscriptome was studied for all the samples from the first experiment and for selected samples for the second experiment (Supplementary Table 3). Two replicates were sequenced for all samples (each replicate was obtained by pooling the RNA extracted from two different cheeses). Ribosomal RNA (rRNA) was depleted by using the Ribo-Zero Magnetic kit (Epicentre, Charlotte, North Carolina) and the mRNA enriched RNA was purified by Agencourt RNAClean XP (Beckman Coulter, Milano, Italy) following the manufacturer’s instruction. Then, library preparation and sample multiplexing were carried out by using the ScriptSeq v2 RNA-Seq Library Preparation Kit (Epicentre, Charlotte, North Carolina) (insert size around 300 bp). The quality of the library was checked by 2100 Bioanalyzer (Agilent Technologies, Palo Alto, California). Sequencing was carried out on a Next Seq 500 Sequencer with the Mid Output Kit (both from Illumina, San Diego, California), yielding 150 bp single-end reads.
Bioinformatics and data analysis
16S rRNA amplicon reads were analyzed by using QIIME 1.9.0 software31, as previously reported30. OTUs defined by a 99% of similarity were picked using the uclust pipeline32 and the representative sequences were submitted to the RDPII classifier33 to obtain the taxonomy assignment and the relative abundance of each OTU using the Greengenes 16S rRNA gene database34.
The whole metatranscriptome data analysis was carried out as follows: raw reads quality (Phred scores) was evaluated by using the FastQC toolkit (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adaptor and primer contamination was eliminated with CutAdapt35. Then, low quality bases (Phred score <20) were trimmed and reads shorter than 60 bp were discarded with the SolexaQA++ software36. Reads were aligned to a reference database by using Bowtie237 in end-to-end, sensitive mode. The database used was built downloading the protein coding portions of the genomes (.ffn files) from the NCBI RefSeq database (ftp://ftp.ncbi.nlm.nih.gov/genomes/ASSEMBLY_BACTERIA/) and from http://patricbrc.org/portal/. The species included were chosen according to the 16S sequencing results and picking species commonly found in food ecosystems (listed in Supplementary Table 6). Since we did not carry out metagenomics and we did not have the genomes of strains directly isolated from those samples, all the available sequenced genomes were included. The concatenated .ffn files were aligned against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database38 version April 2011 by using mblastx39 in order to obtain the functional annotation and the gene taxonomy. The number of reads uniquely mapped to each gene in the database was extracted by using SAMtools40 and normalized according to the library size using custom scripts built under R environment (www.r-project.org). Only genes to which at least 5 reads/sample mapped were kept for subsequent analyses. Statistical analysis and plotting were carried out in R environment. Differential gene expression analysis was done by using the Bioconductor package DESeq41. P-values were adjusted for multiple testing using the Benjamini-Hochberg procedure42 and a false discovery rate (FDR) <0.05 considered as significant. Pairwise Spearman’s correlations between OTUs, KEGG genes, volatile organic compounds and biochemical indices were computed by using the R package psych and the significant ones (FDR < 0.05) were plotted in a correlative network by using Cytoscape v. 2.8.143. Principal Component Analysis (PCA) and Hierarchical Clustering were carried out by using the made4 package in R. All the results are reported as mean values of two replicates.
Extraction and analysis of volatile compounds by SPME-GC/MS
The extraction of volatile compounds was performed according to Lee et al.44. The frozen cheese was finely grated and 25 g were transferred into 100 mL bottle, with 25 mL of deionised water, 50 μL of 2-methyl-3-heptenone (99% purity, Sigma-Aldrich, St. Louis, Missouri) as internal standard (950 mg L−1) and 12.5 g of sodium phosphate (NaH2PO4). The bottle was kept at 50 °C for 10 min in a water bath to melt the cheese. Then melted cheese was magnetically stirred for 20 min at 50 °C to accelerate the equilibrium of the volatile compounds in the headspace. The SPME fiber (50/30 μm thickness divinyl-benzene/carboxen/polydimethylsiloxane, 2 cm; Sigma-Aldrich, St. Louis, Missouri) was inserted through a Teflon septum in the bottle and exposed to sample headspace for 30 min at 40 °C during stirring. The same type of fiber was reported to be highly efficient for cheese aroma analysis45. Volatile compounds were analyzed by GC coupled with a mass spectrometer using a GC/MS Hewlett-Packard 6890N (Agilent Technologies, Palo Alto, California) equipped with a J&W HP-5MS capillary column (30 m × 0,25 mm i.d. × 0,25 μm Film Thickness; J&W Scientific, Folsom, California). Temperature was set at 40 °C for 2 min and increased from 40 to 160 °C at the rate of 6 °C/min and from 160 to 210 °C min−1 at 10 °C min−1. The injector was kept at 250 °C. Helium was used as carrier gas (0.9 mL min−1). Mass spectra were recorded at 70 eV. The source temperature was 230 °C and the interface temperature was 250 °C. Volatile compounds thermal desorption was carried out by exposing the SPME fiber in the injector for 10 min. Compounds identification was performed by comparing retention times and mass spectra with those of pure reference compounds in the same conditions and with those of NIST database. Prior to use, the fiber was conditioned at 270 °C for 1 h. A blank test was carried out before each analysis to prevent the release of undesirable compounds. The quantitative analysis was carried out by normalizing the peak area of each compound respect to the area of the peak of the internal standard. Peak areas were processed by the software Chemstation (Agilent Technologies, Palo Alto, California). The analysis was performed in triplicate.
Significant differences among the different samples were determined for each compound by one-way ANOVA statistical analysis. Tukey’s test was used to discriminate among the means of the variables. Differences with P < 0.05 were considered significant. The data elaboration was carried out using XLStat (version 2009.03.02), an add-in software package for Microsoft Excel (Addinsoft Corp., Paris, France).
Proteolysis and lipolysis analyses
Grated cheese (5 g) was dispersed in 100 mL 0.4 M sodium citrate buffer (pH 8.0) previously heated to 40 °C and homogenised for 1 min with an Ultra-Turrax apparatus (Ultra-Turrax model T25, Ika Labortechnik, Staufen, Germany). The degree of hydrolysis (DH) was determined with the 2,4,6-trinitrobenzene sulfonic acid method, according to Hermanson46 with the modifications reported by Picariello et al.47. For LC-MS analysis, the pH of the citrate soluble fraction was adjusted to 4.6. The resulting suspension was centrifuged for 10 min at 3000 g, the fat layer removed and the insoluble and soluble fractions were separated. The two fractions were then analyzed by LC/ESI-Q/TOF MS/MS using a Q TOF Ultima hybrid mass spectrometer (Waters, Manchester, UK), equipped with an ESI source and operating in positive ion mode, as previously reported48.
The lipolysis index was expressed as the quantity of KOH (in mg) required to neutralize the free fatty acids contained in 1 g of sample. Twenty grams of each frozen sample were finely grated and three grams of the grated cheese were incubated for 30 min with 50 ml of ethanol/diethyl ether (1:1 v/v) while stirring, in order to extract the fat. The suspension was filtered through Watman paper n° 42 and the acidity of the filtrate was measured by titration as described by Dugat-Bony et al.16. Each sample was analysed in triplicate.
Sequences accession numbers
The 16S rRNA and the metatranscriptome sequences are available at the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under accession numbers SRP061555 and SRP061556, respectively.
Additional Information
How to cite this article: De Filippis, F. et al. Metatranscriptomics reveals temperature-driven functional changes in microbiome impacting cheese maturation rate. Sci. Rep. 6, 21871; doi: 10.1038/srep21871 (2016).
References
Erkus, O. et al. Multifactorial diversity sustains microbial community stability. ISME J. 7, 2126–2136 (2013).
Fox, P. F., McSweeney, P. L. H., Cogan, T. M. & Guinee, T. P. Cheese: Chemistry, Physics, Microbiology. (Elsevier Academic Press, 2004).
Montel, M.-C. et al. Traditional cheeses: rich and diverse microbiota with associated benefits. Int. J. Food Microbiol. 177, 136–154 (2014).
De Filippis, F., La Storia, A., Stellato, G., Gatti, M. & Ercolini, D. A selected core microbiome drives the early stages of three popular Italian cheese manufactures. PLoS ONE 9(2), e89680 (2014).
Ercolini, D., Frisso, G., Mauriello, G., Salvatore, F. & Coppola, S. Microbial diversity in natural whey cultures used for the production of Caciocavallo Silano PDO Cheese. Int. J. Food Microbiol. 124, 164–170 (2008).
Coppola, R., Succi, M., Sorrentino, E., Iorizzo, M. & Grazia, L. Survey of lactic acid bacteria during the ripening of Caciocavallo cheese produced in Molise. Lait 83, 211–222 (2003).
De Pasquale, I., Di Cagno, R., Buchin, S., De Angelis, M. & Gobbetti, M. Microbial ecology dynamics reveal a succession in the core microbiota involved in the ripening of pasta filata Caciocavallo Pugliese cheese. Appl. Environ. Microbiol. 80, 6243–6255 (2014).
Di Cagno, R., Quinto, M., Corsetti, A., Minervini, F. & Gobbetti, M. Assessing the proteolytic and lipolytic activities of single strains of mesophilic lactobacilli as adjunct cultures using a Caciotta cheese model system. Int. Dairy J. 16, 119–130 (2006).
Fröhlich-Wyder, M.-T., Guggisberg, D., Badertscher, R., Wechsler, D., Wittwer, A. & Irmler, S. The effect of Lactobacillus buchneri and Lactobacillus parabuchneri on the eye formation of semi-hard cheese. Int. Dairy J. 33, 120–128 (2013).
Gobbetti, M., Morea, M., Baruzzi, F., Corbo, M. R., Matarante, A. & Considine, T. Microbiological, compositional, biochemical and textural characterisation of Caciocavallo Pugliese cheese during ripening. Int. Dairy J. 12, 511–523 (2002).
Piraino, P., Zotta, T., Ricciardi, A. & Parente, E. Discrimination of commercial Caciocavallo cheeses on the basis of the diversity of lactic microflora and primary proteolysis. Int. Dairy J. 15, 1138–1149 (2005).
Mauriello, G., Moio, L., Genovese, A. & Ercolini, D. Relationships between flavoring capabilities, bacterial composition and geographical origin of natural whey cultures used for traditional water-buffalo mozzarella cheese manufacture. J. Dairy Sci. 86, 486–497 (2003).
Smit, G., Smit, B. A. & Engels, W. J. Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese products. FEMS Microbiol. Rev. 29, 591–610 (2005).
O’Sullivan, D. J., Giblin, L., McSweeney, P. L. H., Sheehan, J. J. & Cotter, P. D. Nucleic acid-based approaches to investigate microbial-related cheese quality defects. Front. Microbiol. 4, 1 (2013).
Cocolin, L. & Ecolini, D. Zooming into food-associated microbial consortia: a “cultural” evolution. Curr. Opin. Food Sci. 2, 43–50 (2015).
Dugat-Bony, E. et al. Overview of a surface-ripened cheese community functioning by meta-omics analyses. PLoS ONE 10(4), e0124360 (2015).
Lessard, M.-H., Viel, C., Boyle, B., St-Gelais, D. & Labrie, S. Metatranscriptome analysis of fungal strains Penicillium camemberti and Geotrichum candidum reveal cheese matrix breakdown and potential development of sensory properties of ripened Camembert-type cheese. BMC Genomics 15, 235 (2014).
Wolfe, B. E. & Dutton, R. J. Fermented food as experimentally tractable microbial ecosystems. Cell 161, 49–55 (2015).
O’Sullivan, D. J., Cotter, P. D., O’Sullivan, O., Giblin, L., McSweeney, P. L. H. & Sheehan, J. J. Temporal and spatial differences in microbial composition during the manufacture of a continental-type cheese. Appl. Environ. Microbiol. 81, 2525–2533 (2015).
Wolfe, B. E., Button, J. E., Santarelli, M. & Dutton, R. J. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 158, 422–433 (2014).
Law, J. & Haandrikman, A. Proteolytic enzymes of lactic acid bacteria. Int. Dairy J. 7, 1–11 (1997).
Liu, M., Bayjanov, J. R., Renckens, B., Nauta, A. & Siezen, R. J. The proteolytic system of lactic acid bacteria revisited: a genomic comparison. BMC Genomics 11, 36 (2010).
Savijoki, K., Ingmer, H. & Varmanen, P. Proteolytic systems of lactic acid bacteria. Appl. Microbiol. Biotechnol. 71, 394–406 (2006).
Morea, M., Matarante, A., Di Cagno, R., Baruzzi, F. & Minervini, F. Contribution of autochtonous non-starter lactobacilli to proteolysis in Caciocavallo Pugliese cheese. Int. Dairy J. 17, 525–534 (2007).
Di Cagno, R., De Pasquale, I., De Angelis, M. & Gobbetti, M. Accelerated ripening of Caciocavallo Pugliese cheese with attenuated adjuncts of selected nonstarter lactobacilli. J. Dairy Sci. 95, 4784–4795 (2012).
Weimer, B. C. Improving the Flavour of Cheese (Elsevier, 2007).
Ganesan, B., Seefeldt, K. & Weimer, B. C. Monocarboxylic acid production by lactococci and lactobacilli. Int. Dairy J. 14, 237–246 (2004).
Marillay, M. & Casey, M. G. Flavours of cheese products: metabolic pathways, analytical tools and identification of producing strains. Int. J. Food Microbiol. 90, 139–159 (2004).
Ercolini, D., De Filippis, F., La Storia, A. & Iacono, M. “Remake” by high-throughput sequencing of the microbiota involved in the production of water buffalo mozzarella cheese. Appl. Environ. Microbiol. 78, 8142–8145 (2012).
De Filippis, F. et al. The same microbiota and a potentially discriminant metabolome in the saliva of omnivore, ovo-lacto-vegetarian and vegan individuals. PLoS ONE 9(11), e112373 (2014).
Caporaso J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618 (2012).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. The EMBnet Journal 17, 120–112 (2011).
Cox, M. P., Peterson, D. A. & Biggs, P. J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11, 485 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 (2014).
Davis, C. et al. mBLAST: keeping up with the sequencing explosion for (meta) genome analysis. J. Data Mining Genomics Proteomics 4, 135 (2013).
Li, H. et al. The sequence alignment/map format and samtool. Bioinformatics 25, 2078–2079 (2009).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B. Stat. Methodol. 57, 289–300 (1995).
Shannon, P. et al. Cytoscape: A Software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
Lee, J. H., Diono, R., Kim, G. Y. & Min, D. B. Optimization of solid phase microextraction analysis for the headspace volatile compounds of Parmesan cheese. J. Agric. Food Chem. 51, 1136–1140 (2003).
Gioacchini, A. M., De Santi, M., Guescini, M., Brandi, G. & Stocchi, V. Characterization of the volatile organic compounds of Italian “Fossa” cheese by solid‐phase microextraction gas chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 24, 3405–3412 (2010).
Hermanson, G. T. Bioconjugate Techniques (Elsevier Academic Press, 1996).
Picariello, G. et al. Role of intestinal brush border peptidases in the simulated digestion of milk proteins. Mol. Nutr. Food Res. 59, 948–956 (2015).
Ferranti, P., Nasi, A., Bruno, M., Basile, A., Serpe, L. & Gallo, P. A peptidomic approach for monitoring and characterizing peptide cyanotoxins produced in Italian lakes by matrix-assisted laser desorption/ionization and quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 25, 1173–1183 (2011).
Acknowledgements
F.D.F. was supported by a grant from Regione Campania within the program “POR CAMPANIA FSE 2007/2013” - project CARINA (Safety sustainability and competitiveness of the agro-food production in Campania) – CUP B25B09000080007. We would like to thank the dairy Campolongo S.r.l., Montesano Sulla Marcellana – Italy for providing the samples for this study.
Author information
Authors and Affiliations
Contributions
F.D.F. performed the 16S and metatranscriptomics experiments and the data analysis and wrote the manuscript. D.E. designed the study and contributed to manuscript preparation. J.A.G. contributed to data analysis and manuscript preparation. A.G. and P.F. performed the analysis of metabolome.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
De Filippis, F., Genovese, A., Ferranti, P. et al. Metatranscriptomics reveals temperature-driven functional changes in microbiome impacting cheese maturation rate. Sci Rep 6, 21871 (2016). https://doi.org/10.1038/srep21871
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21871
This article is cited by
-
Effect of lactic acid bacteria on the structure and potential function of the microbial community of Nongxiangxing Daqu
Biotechnology Letters (2023)
-
Microbial diversity in jiuqu and its fermentation features: saccharification, alcohol fermentation and flavors generation
Applied Microbiology and Biotechnology (2023)
-
The effects of an innovative pulping technique of synchronously pulping and gelatinizing treatment on raw materials properties, oenological parameters, fermentation process, and flavor characteristics of glutinous rice wine
Food Science and Biotechnology (2022)
-
Meta-analysis of cheese microbiomes highlights contributions to multiple aspects of quality
Nature Food (2020)
-
Assessment of overall microbial community shift during Cheddar cheese production from raw milk to aging
Applied Microbiology and Biotechnology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
