
Abstract
Pathology in Parkinson’s disease is linked to self-association of α-Synuclein (αS) into pathogenic oligomeric species and highly ordered amyloid fibrils. Developing effective therapeutic strategies against this debilitating disease is critical and βS, a pre-synaptic protein that co-localizes with αS, can act as an inhibitor of αS assembly. Despite the potential importance of βS as an inhibitor of αS, the nature, location and specificity of the molecular interactions between these two proteins is unknown. Here we use NMR paramagnetic relaxation enhancement experiments, to demonstrate that βS interacts directly with αS in a transient dimer complex with high specificity and weak affinity. Inhibition of αS by βS arises from transient αS/βS heterodimer species that exist primarily in head- to- tail configurations while αS aggregation arises from a more heterogeneous and weaker range of transient interactions that include both head-to-head and head-to-tail configurations. Our results highlight that intrinsically disordered proteins can interact directly with one another at low affinity and that the transient interactions that drive inhibition versus aggregation are distinct by virtue of their plasticity and specificity.
Similar content being viewed by others
Introduction
Protein aggregation is the origin of a wide variety of human neurodegenerative diseases including Parkinson’s, Alzheimer’s, Huntington’s and Creutzfeldt-Jakob disease1,2. Parkinson’s disease (PD), a highly debilitating illness, is the second most prevalent of the late onset neurodegenerative diseases and affects as many as 6 million people worldwide3. Αs life expectancy continues to increase, neurodegenerative diseases like Parkinson’s are becoming increasingly common and a threat to global public health. The diagnostic hallmark of PD is a deposit called a Lewy body that is primarily composed of the pre-synaptic intrinsically disordered protein α-synuclein (αS). Fibril formation of αS is also implicated in other neurodegenerative diseases, including multiple system atrophy and dementia with Lewy bodies, referred to as synucleinopathies. Although the function of αS is not clearly defined it is thought to be involved in promoting SNARE complex assembly4,5, in regulation of the synaptic vesicle pool6,7 and in remodeling membranes8. The origin of αS pathology in neurodegenerative disease is clearly linked to the self-association of the intrinsically disordered αS monomers into pathogenic oligomeric species and highly ordered amyloid fibrils.
One approach to developing effective therapeutic strategies against this debilitating disease is to identify inhibitors of αS aggregation. Small molecule inhibitors of αS have been proposed9,10,11,12,13. In addition, proteins such as heat shock proteins (Hsp40, Hsp70, Hsp90, αB-crystallin)14,15 and the intrinsically disordered β-synuclein (βS), a homologue of αS with which it co-localizes have been shown to interfere with αS assembly. A number of studies have established a neuroprotective role for βS16,17,18,19,20,21,22,23,24. Masliah and co-workers have shown that βS is expressed at similar levels as to αS in the central nervous system. However the ratio of βS to αS at the mRNA level is significantly decreased in diseased brains, suggesting a regulatory role within the synuclein family24. In vivo it has been shown that over-expression of αS with βS in mouse models significantly decreases the number of plaques formed25 and that intracerebral injection of the lenti-βS virus reduces the formation of αS inclusions in transgenic mice26. In vitro, it has been shown that the presence of βS with αS slows its aggregation16,17,18,25,26,27. Despite the fact that βS has a very similar sequence to αS, it does not form fibrils on its own16,28,29,30, but may form aggregates whose toxicity is debated31,32. The in vivo data clearly suggest that βS plays an important regulatory role in inhibition of αS pathology but at this stage there is no molecular information about the nature, location and specificity of the protein-protein interactions that initiate the inhibition of αS by βS.
To understand the mechanism by which the intrinsically disordered protein βS interacts with αS, we use NMR to map the monomer-monomer interactions that lead to inhibition or promotion of aggregation. Despite the importance of these interactions, the molecular details are extremely difficult to obtain due to their transient nature and low population. Paramagnetic relaxation enhancement experiments (PRE) offer an excellent tool for characterization of weak and transient interactions because they are able to probe states that exist at low populations (even 0.5–5%) and exhibit short life times (250–500 μs)33,34. Here we use inter-chain NMR PRE experiments to identify and characterize weak transient complexes of αS and βS35. We show that αS homo-dimers sample a heterogeneous range of population distributions, including head- to- head and head- to- tail configurations, while αS/βS hetero-dimers exist primarily in head- to- tail configurations.
NMR inter-chain PRE titration experiments previously used on folded proteins36 are applied here to intrinsically disordered proteins. These experiments allow us to obtain residue specific dissociation constants to inform us about the specificity and affinity of dimer interactions in the different regions of the transient disordered complexes. Our results show that the hetero-dimer transient head- to- tail interactions between αS and βS are approximately 5 times stronger than the interactions observed in the homo-dimer αS species suggesting that these αS/βS interactions create a kinetic trap which delays or inhibits the formation of αS fibrils. The novel insight presented in this paper not only defines residue specific contacts between two intrinsically disordered proteins but also links the homo and hetero-complexes with distinct pathways that lead to aggregation versus inhibition.
Results
βS inhibits αS fibril formation in a dose dependent manner
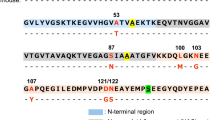
αS and βS are part of the synuclein family. These can be described as: the N-terminus that contains KTKXGV repeats and forms helices at membranes37, the non-amyloid-βS component (NAC) region and the highly acidic and solubilizing C-terminus (Fig. 1). αS and βS have similar sequences, particularly at the N-terminus, but very different fibrillation and oligomerization properties (Fig. 1). The N-terminus for all synucleins is highly conserved, with only 6 substitutions between αS and βS sequences. In contrast, the C-terminus is the least conserved region with more prolines and more negatively charged residues with a net charge of -12 in αS and -15 in βS. βS has an 11 residue deletion in the NAC region that was thought to be important in preventing fibril formation but substitution of this region into βS does not recover full fibrillation potential of αS28,29.

Sequence alignment of αS and βS shows high similarity between proteins.
Sequence differences are color-coded according to the hydrophobicity index shown in the bottom right (red-hydrophobic, blue-hydrophilic). Identical residues are shown by dots and deletions are shown by dashes. The line above the sequence shows the N-(blue), NAC (grey) and C-terminal regions (pink).
Similarly to αS it has been established that the physiological form of βS and its pathological mutants in vivo are N-terminally acetylated38. All experiments in this study are performed on the acetylated forms of the protein that we will refer to as αS and βS. All previous characterization of βS was performed on non-acetylated protein16,17,28,39, therefore we use NMR and other biophysical approaches here to determine whether N-terminal acetylation affects the conformation or oligomerization state of the protein. NMR and other biophysical techniques including dynamic light scattering (DLS) and circular dichroism (CD) show that acetylated βS, similarly to acetylated αS, is primarily monomeric and unfolded (Supplementary Fig. 1 & 2). Secondary structure propensities indicate the formation of a transient N-terminal helix relative to the non-acetylated form of βS similarly to αS (Supplementary Fig. 1 & 2). In addition, the C-terminus of acetylated βS is more extended that the C-terminus of acetylated αS consistent with previous results on non-acetylated protein40. Electrospray ionization mass spectroscopy (ESI-MS) experiments (Supplementary Fig. 1) show that acetylated βS can populate an extended and a compact form with a higher population of extended conformation relative to acetylated αS.
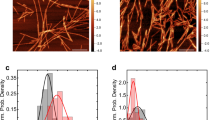
Addition of acetylated βS to acetylated αS inhibits fibril formation in a dose dependent manner, consistent with previous findings for the non-acetylated forms of the proteins (Fig. 2A)16. As the concentration of βS is increased we observe a significant change in the rate of the elongation phase and in the total ThT intensity as well as a small change in the lag phase. Previous work has shown that ThT can be used as a semiquantitative method to estimate relative amounts of fibril formed, although caution should be applied in interpreting ThT intensities41,42. In this case, the inhibition of fibril formation as shown by the ThT experiments is supported by Transmission Electron Microscopy (TEM) data which shows significant changes in fibril morphology of αS/βS relative to fibrils of αS alone (Fig. 2B–D). βS does not form fibrils and the αS/βS mixture forms significantly fewer and shorter fibrils than αS alone as seen in TEM (Fig. 2C,D).

Aggregation inhibiton of αS by βS as monitored by ThT and TEM.
(A) ThT fibrillation assay curves of: αS alone (blue), βS alone (blue), αS+βS 1:1 mixture (violet), αS+βS 1:5 mixture (magenta) Experimenal conditions: 10 mM MES, pH 6, 70 μM protein. The experiment was performed in triplicate at 37 °C with shaking with teflon beads and average values were plotted with associated error bars. The buffer conditions matched the conditions of NMR PRE expreriments. Negatively stained electron micrographs (scale 100 nm) of end products of the ThT fibrillation assay of: (B) αS (C) βS (D) αS co-incubated with βS, ratio 1:1.
Mapping of residue specific transient interactions in αS/αS homo- and αS/βS hetero-dimer complexes using NMR inter-chain PRE experiments
ThT fluorescence experiments and TEM data (Fig. 2) have established that βS alters the aggregation kinetics of αS, however there has been no evidence to date of a direct interaction between these proteins. To determine the existence of and to characterize transient inter-chain interactions between homo-dimers (αS/αS, βS/βS) and hetero-dimers (αS/βS, βS/αS), inter-chain NMR paramagnetic relaxation enhancement experiments were performed. In this experiment, NMR blind 14N-MTSL labeled protein is mixed with NMR visible (15N) unmodified protein and broadening of signal is limited to residues on the NMR visible chain that interact with the MTSL labels on the NMR blind chain (Supplementary Fig. 3). Signal broadening therefore reflects interactions between the NMR blind and NMR visible protein33,34,35,43.
NMR PRE experiments were performed on all four combinations of possible homo- and hetero-dimers (αS15NαSMTSL, αS15NβSMTSL, βS15NαS MTSL, βS15NβSMTSL) to determine whether there is evidence for direct residue specific transient interactions and to establish the location of the interactions. The results of Fig. 3 are striking as they show immediately that the four combinations of possible hetero and homo-dimers have different transient interaction profiles (Fig. 3 and Supplementary Fig. 4). There is evidence for interactions between the homo-dimer complexes of αS and the hetero-dimer complexes of αS and βS; interactions between βS are essentially non-existent. We have introduced four spin labels along the sequence at positions 11, 44, 90 and 132 for αS and 11, 44, 80, 134 for βS to probe interactions in the N, NAC and C-terminal regions. Experimental results are presented as heat maps (Fig. 3A–D), where each strip shows color-coded values of residue-specific inter-chain paramagnetic relaxation enhancement rates (PRE rate-Γ) induced by the proximity of the MTSL label to another chain. Strong interactions (>12 Hz) are defined relative to the lack of interactions otherwise observed in the βS homo-dimer contact maps. Under the conditions of the experiment, αS and βS do not form fibrils or oligomers; therefore dimer detection arises as a result of low populations of dimers existing in equilibrium with the monomer precursor (for αS lack of higher order species in the sample has been confirmed by ESI-IMS-MS experiments44 and for hetero-species this has been confirmed with ESI-MS.

Contact maps of αS/αS homo-dimers and αS/βS hetero-dimers show distinctive interaction profiles.
Contact maps of transient dimers, shown as heat maps where each strip represents the color coded value of the residue-specific inter-chain paramagnetic relaxation enhancement rate (PRE rate-HNΓ2) induced by the proximity of the MTSL label to the residues in the indicated protein. Contact maps (A–D): figures A-D show four heat maps for all possible permutations of the spin label and NMR detectable chains of αS and βS: (A) 14N-αS-MTSL/15N-αS, (B) 14N-αS-MTSL/15N-βS, (C) 14N-βS-MTSL/15N-αS, (D) 14N-βS-MTSL/15N-βS. Contact maps show the PRE values colored in accordance with the legend; residues that do not exhibit interactions are colored blue and interactions higher than 12 Hz are colored red. Each strip on the contact maps corresponds to the spin label (y-axis). In one strip there are bins which correspond to the residue number on the NMR visible chain (x-axis). (E,F) Schematic representations of possible interactions of homo αS/ αS and hetero- αS/βS synuclein dimers. (E) αS/αS corresponds to interactions from the contact map (A,F) αS/βS corresponds to contact map (B,C); no homo-dimer for βS is shown as we observe no inter-chain interactions between βS chains. HS in the schematic stands for interactive “Hot Spot”. The hot spot size is defined based on the interactivity of the regions as shown in Supplementary Fig. 4A. Hot spot encompasses residues 38–45.
αS populates a heterogeneous range of transient complexes while αS/βS hetero-complexes sample primarily head-to–tail non-propagating interactions
The heat map for the αS/αS homo-dimer shows strong interactions between the N-terminal αS-MTSL labeled positions A11 and T44 with the N-terminal region 36 to 44 and the C-terminal region 124 to 140 (Fig. 3A). Earlier studies by our group35 on αS showed transient inter-chain interactions between the N- and C- termini (N-C), but by increasing the number and positions of the MTSL spin labels we now observe new inter-chain interactions between the N-termini showing that αS can populate multiple dimer configurations. In noticeable contrast, the αS/βS hetero-dimers show strong interactions between the N-terminal αS-MTSL labeled positions A11 and T44 and the C-terminus from residues 105 to 134 and extremely weak N-N terminal interactions between 37 and 41 (Fig. 3B). According to the heat map, the hetero-dimer interactions between the N-terminus of αS and the C-terminus of βS appear to be more extensive and stronger (residues 105-134 compared to residues 124-140 in αS/αS homo-dimers) (Fig. 3A–C) than those in αS. βS shows extremely minimal interactions with itself supporting the view that βS does not form fibrils (Fig. 3D).
A schematic representation of the dominant homo and hetero-dimer interactions shows that both N-N and N-C configurations are sampled by the αS homo-dimers while only N-C terminal interactions are sampled in the αS/βS hetero-dimers (Fig. 3E,F). The favorable interactions of the N-terminal hydrophobic region of αS with itself (N-N) and with the C-terminus of αS and βS suggest that they act as an aggregation initiation region. The highly interactive N-terminal region encompassing residues 38- 45 is referred to as ‘hot spot’ region (Suppl. 4A). In the αS homo-complexes, the interactions detected by N- and C-terminal probes show symmetry, implying that the interactions we observe are not experimental artifacts. The NMR PRE data show that αS homo-dimers can sample a heterogeneous range of populations, including head- to- head and head- to- tail configurations while αS/βS hetero-dimers sample only head- to- tail dimers. This suggests that the hetero-dimers have a more limited range of conformational preferences for the dimer species.
Aggregation versus inhibition of αS by βS is due to a balance between specificity and affinity of transient interactions
The inter-chain NMR PRE experiments described above are powerful as they provide us with direct evidence for the existence of transient interactions and allow us to pinpoint the specific residues involved in these encounter complexes, however the specificity and affinity of the interactions remains unknown. We extend an earlier approach36,45 designed for folded proteins to transient encounter complexes of IDPs. We obtain residue-specific equilibrium dissociation constants (KD) by performing titrations of 15N labeled αS with MTSL-labeled 14N-αS. KDs are obtained by fitting the intermolecular transverse 1H relaxation rates to a titration curve (see methods) (Fig. 4).

Residue specific binding affinities for transient αS/βS hetero-complexes show higher specificity and affinity than αS/αS homo-complexes.
(A–C) Representative examples of titration curves arising from PRE titration profiles from αS-44-MTSL to 15N-αS and 15N-βS. Illustration of how the KD is obtained from fitting the PRE titration data as a function of the molar ratio of 14N-αS-T44C-MTSL labeled to 15N-αS (A,B) and to 15N-βS (C). (A) αS residues 38–41 show a linear dependence between PRE values and concentration indicating non-specific interactions at these sites, (Residues colors: 38-black, 39- blue, 40-pink, 41-green), (B) αS residues 125–140 exhibit non-linear titration curves indicating specific interactions. (Residues colors: 125-orange, 130-blue, 135-pink, 137-green, 139-black), (C) βS residues 109–134 interact strongly and more uniformly with αS-44-MTSL (Residues colors: 115-green, 120-orange, 121-blue, 129-pink, 131-black). Shaded areas indicate the range of possible KD values. Titration profiles for αS/βS show a higher degree of saturation consistent with more specificity and higher affinity than those for αS/ αS (D,E) 3D representation of titration curves in the C-terminus (D) 15N-αS C-terminus titrated with T44C-MTSL labeled 14N-αS (E) 15N-βS C-terminus titrated with T44C-MTSL labeled 14N-αS titration. The x-axis shows the C-terminal residues of both αS and βS after sequence alignment, the z- axis depicts the PRE values, the y- axis the ratio of T44C-MTSL labeled 14N-αS to 15N sample concentration. The surface is colored using a rainbow palette, where low HNΓ2 values are red and the highest values are purple (according to legend below the plot).
NMR PRE titration experiments and data analysis is complex as KDs are anticipated to be weak due to the disordered nature of the monomers. The titration curves are grouped according to their profiles and three different types of patterns emerge (Fig. 4A–C). Representative examples of titration curves arising from interactions between αS-44-MTSL and residues 38–41 of the N-terminus (Fig. 4A) and residues 125–140 of the C-terminus (Fig. 4B), show that they are distinct from one another. The titration curves for the N-N interactions show a linear dependence between PRE values and concentration indicating non-specific interactions at this site, while interactions with the C-terminal 125–140 exhibit non-linear titration curves suggesting specific interactions. The range of KD values in this group is between KD ~ 500 μM (range 90–1200 μM) using the data analysis described in methods. While we observe both non-specific (N-N) (Fig. 4A) and specific interactions (N-C) (Fig. 4B) in homo-αS complexes, the hetero-complexes αS/βS (Fig. 4C) have only specific N-C interactions. The titration profiles of αS-44-MTSL with βS in the region 115–134 have a narrower range of KD values (KD ~ 100 μM, range 40–350 μM) than those associated with the KD values of αS-44-MTSL αS with its own C-terminus (KD ~ 500 μM, range 90–1200 μM) suggesting more uniform behavior across this region and higher specificity and affinity by approximately 5 fold (Supplementary Tables 1&2). The differences between the strengths of the interactions and the range of residues over which the interactions are occurring is seen clearly in the 3D plots where the interaction regions in αS are more rugged while the interactions between αS and βS are smoother, more uniform and extend over a wider range of residues (Fig. 4D,E).
Discussion
βS plays a role in the inhibition of αS aggregation but the mechanism by which this occurs and the stage in the aggregation pathway at which βS first interacts with αS has been unknown. We demonstrate that the monomer species of αS and βS interact directly with one another at specific sites suggesting that inhibition may begin at the very earliest stages of the fibril formation process. The molecular interactions and affinities obtained in the NMR PRE experiments described here support the view that early stages of aggregation versus inhibition may be due to a balance between conformational heterogeneity, specificity and affinity. Early stages of aggregation in αS may be promoted by sampling or searching conformational structures with weak transient affinities, including both non-specific head- to- head and weak specific head- to- tail dimers (Fig. 5A). In contrast early stages of inhibition may be favored by sampling only head- to- tail interactions with higher affinity and specificity within the αS/βS hetero-complex (Fig. 5B).

Schematic model of αS/αS and αS/βS transient interactions suggest a new molecular view of inhibition routes to αS aggregation.
(A) αS/αS homo-dimer interactions: head-to-head aggregation prone interactions at residues 36–44 co-exist with head-to-tail inhibitory interactions at residues 125–140. (B) αS/βS hetero-dimer interactions: head- to- tail inhibitory interactions span a broader range of C-terminal residues 115–134 with higher affinity. (C) No interactions are observed between monomer chains of βS. Protein chains are color-coded according to the interactivity of the regions as shown in Supplementary Fig. 4. The NMR data show that αS homo-dimers can sample a heterogeneous range of poulations including N-to-N and N- to C-terminal configurations while αS/βS hetero-dimers sample only N- to C- terminal configurations with higher affinity and specificity. In light of the fact that the fibril form of αS forms β-strands that assemble into in register parallel β-sheets this would suggest that the N-C terminal dimers would have to undergo conformational rearrangement to reach the final fibril form, thereby delaying or inhibiting the kinetics of fibril formation.
Head- to- tail interactions exist in both the homo- and hetero-dimer complexes, which strongly suggests that they may play a regulatory role on αS folding or misfolding. In light of the fact that the fibril state of αS assembles into in-register parallel cross-β-structure46,47, the NMR data suggests that head-to-tail αS/βS dimers would have to undergo conformational rearrangement to reach the final fibril form. This may thereby delay or inhibit the kinetics of fibril formation. We propose that the αS homo-complexes are more aggregation prone than αS/βS complexes for two reasons: first, αS can sample head- to- head interactions that are potentially aggregating promoting while αS/βS does not sample these; second, the non-propagating head- to- tail αS/αS complex has weaker, lower affinity interactions relative to the αS/βS complex suggesting that the conformational rearrangement required for fibril formation may be more facile for the αS/αS homo-complex.
Despite the fact that both the αS/αS homo-dimer and the αS/βS hetero-dimer sample head- to –tail interactions, there are notable differences in terms of the strength of the interactions and the range over which the interactions extend. For homodimers the interaction of the αS “hot spot” with the C-terminus of αS extends over ~15 residues, while the interaction of the αS “hot spot” with the C-terminus of βS extends over a broader range of residues from 105 to 134. In both cases the dimers are flexible and are able to probe a big surface area, possibly adopting multiple dimer conformations. In addition, the αS and βS C-termini are highly negatively charged and contain aromatic and hydrophobic residues suggesting that initial interactions are mediated by electrostatics, which are anchored and stabilized through the hydrophobic and aromatic interactions.
Using NMR PRE titration experiments we demonstrate that interactions between the C-terminus of βS and the N-terminus of αS are approximately 5 times stronger and more extensive than the interactions of the C-terminus of αS with its own N-terminus. This increased specificity and affinity may be attributed to two factors: a higher content of negative C-terminal charges thereby enhancing electrostatic interactions and a higher proportion of proline residues thereby altering the conformational ensembles sampled by the C-terminus. NMR and ESI data indicate that the C-terminal of βS has a higher population of more extended species (Supplementary Fig. 1) and may therefore provide a more accessible surface area for interactions with the N-terminus “hot spot’ of αS. These differences suggest that small changes in the binding surface, even in these highly dynamic IDP complexes, can lead to substantial changes in interactivity that can modulate the pathway of protein aggregation versus inhibition.
In conclusion, our studies highlight that transient and weak interactions are important for protein recognition pathways of IDPs that lead to diseases such as amyloidosis where the proteins self-associate and propagate to highly ordered fibrils. Work by Radford et al.48 have shown that weak interactions are also important for folded proteins in directing aggregation versus inhibition. By performing NMR inter-chain PRE titration experiments we have identified and characterized the strength and affinity of transient interactions between αS and βS, both IDPs. As IDPs are highly flexible, binding affinities are likely to behave in a non-cooperative manner across the protein49. Our data support this view and highlight the variable nature of the binding affinities that result in aggregation promoting versus aggregation inhibiting configurations.
Knowledge about the distinct dissociation constants of different interaction regions provides a new framework for thinking about therapeutic intervention by providing direct information about which regions to target for small molecule intervention and about the conformational features that may be most effective at intervention. There have been some small molecule inhibitors that have targeted the N-terminus of αS but now based on our detailed molecular understanding of αS/βS interactions we can optimize the surface interactions to design novel inhibitors10,11,12,50,51,52. In addition, the powerful methods used here to identify the early stages of interaction of αS with βS can be extended to design inhibitors with biologics and can be applied even more widely to study other cross amyloid interactions, such as those between αS and amyloid- β-protein, or αS and tau, that have been shown to play a critical role in cross-seeding in neurodegenerative disease48,53,54,55,56.
Methods
Mutagenesis, expression and purification
Cysteine mutants of αS (A11C, T44C) and βS (A11C, T44C, A80C, A134C), were prepared by site-directed mutagenesis using AccuPrime pfx from Invitrogen. To obtain N-terminal acetylated forms of αS and βS proteins, co-expression with the NatB plasmid N-Acetyltransferase B was performed, as described previously44. Protein purification was performed according to previous protocols57. Similarly, MTSL spin label conjugation to cysteine mutants was performed using previously established protocols35.
PRE experiments/controls
All NMR PRE experiments were performed in 10mM MES, pH 6, without addition of salt and with 10% D2O required for NMR experiments. NMR inter-chain PRE experiments were performed by mixing NMR blind 14N-MTSL labeled protein with NMR visible 15N unmodified protein35. Samples were prepared as follows: lyophilized samples of 14N-MTSL-cysteine mutants or 15N non-modified proteins (αS or βS) were separately dissolved. Samples were passed through a 100 kDa filter to remove higher order oligomers and then concentrated using 3 kDa filters to be able to dilute the sample to a final concentration of 250 uM. Low sample concentration was chosen to minimize non-specific interactions. The total sample concentration was 500 uM, with 250 uM non-modified 15N protein and 250 μM 14N-MTSL labeled protein. All combinations of proteins were mixed in order to see all possible interactions. Diamagnetic samples were prepared by reducing samples with 10× excess of Ascorbic Acid and 5× buffer exchange using 3kDa cutoff filters from Millipore Inc. All the controls have similar patterns and are in the range of experimental error. Additionally, the pattern for the reduced diamagnetic control is consistent with the pattern for the mixture of 14N and 15N non-modified samples. The 1H-15N HSQC of the cysteine mutants with the reduced spin label did not disrupt the HSQC pattern of αS and βS.
All 1H-R2 measurements of paramagnetic and diamagnetic (reduced) samples were acquired on a 600 MHz Varian at 15 °C using previously published pulse sequence and protocols33,35. The inter-chain paramagnetic relaxation enhancement rate (PRE rate–Γ2) is the residue specific difference of the 1H-R2 values of the paramagnetic and diamagnetic samples. Increased 1H-R2 relaxation rates on the 15N visible chain indicates that the NMR blind 14N -MTSL labeled protein is in the proximity of specific residues in the NMR 15N labeled visible chain. Para- and diamagnetic 1H-R2 were analyzed and processed using nmrpipe58 and sparky59. For all experiments 10 relaxation delays were used: 12, 32, 104, 12, 124, 64, 48, 94, 64, 20 ms. Two data points (12 ms, 64 ms) were repeated in the experiment to obtain good statistics for the error analysis. Errors of Γ2 were calculated using error propagation and errors were below 2 Hz. All of the interactions that were considered significant were at least 2 times higher than the mean value and all of them were higher than the 3rd quartile and at least 8Hz.
NMR PRE titration experiments
For NMR PRE titration experiments we used protocols described before36,45. Spin label sample concentrations were reduced to low volume >32 uL and were added to 350uL of 250uM 15N sample of either αS or βS; the changes in the sample volume were less that 10% of the overall sample volume. For αS titrations we used the following ratio of 14N- αS-44-MTSL labeled samples to 15N NMR visible samples: 0, 0.25, 0.5, 0.75, 1, 1.5 and in the case of βS we went up to ratio of 2. αS in this ratio exhibited shifts in the HSQC spectra, thus we removed this point from the analysis. We ran 6 data points ranging from 12–125 ms with the first point repeated twice for statistics. The PRE profiles during the titration did not show significant contributions from the non-interactive regions. The relaxation rates for the non-interactive NAC region for the titration ratio 15N/14N equal to 1.5 are well below 8 Hz.
NMR PRE titration fitting and analysis
Titration curves of 14N-MTSL labeled protein with 15N protein were fit using equation (1):

where x is the concentration of 14N-αS (T44C-MTSL) in solution, Γ2free–represents paramagnetic relaxation enhancement for free protein and Γ2bound represents the maximum observed saturation value. The fitting scheme was based on the papers by Bax45 and Clore36. We used a nonlinear regression model with three fitting parameters Γ2,free, Γ2bound and Kd. The χ-squared statistic that measures the difference between the observed and predicted increase in 1HNΓ2app was optimized using the R statistics package minpack.lm, via the Levenberg-Marquardt algorithm60. The resulting fit was robust to small changes in Γ2free, KD and Γ2bound and provided us with residue specific KD values between 14N-αS-44-MTSL and 15N-αS or 15N-βS. The Kd calculations were performed for residues whose 5th titration point had PRE values higher that 15 Hz (Supplementary table 1&2). Results are summarized in the supplementary tables 1&2.
Thioflavin T (ThT) aggregation assays
The experimental set up for measuring time dependent fibril formation with ThT has been previously described57. The conditions used for the ThT assay match the conditions of the NMR PRE experiments described earlier. 5–10 mg of lyophilized αS and βS was dissolved in 10 mM MES, pH 6 and centrifuged for 10 min. at 14000 rpm to remove big oligomers and purified using size exclusion chromatography (Superdex 75 GL 10/300, from GE Healthcare Life Sciences). Protein was subsequently concentrated using 3kDa centrifugal units (Millipore Inc). The final concentration of αS and/or βS was 70 μM. Addition of βS to αS was performed in multiples of 70 μM. ThT fibrillation rates of the following samples were measured: (1) αS alone (2) βS alone; (3) ThT fluorescence of 1:1 the mixture αS + βS and (4) ThT fluorescence of 1:5 the mixture of αS+5xβS. Each condition was repeated 3 times in 37 °C, with linear shaking in the presence of the Teflon beads. For each sample 3 curves are plotted on the figure to show the variability of the aggregation profiles.
Additional Information
How to cite this article: Janowska, M. K. et al. Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein. Sci. Rep. 5, 15164; doi: 10.1038/srep15164 (2015).
References
Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4, 49–60 (2003).
Uversky, V. N., Oldfield, C. J. & Dunker, A. K. Intrinsically disordered proteins in human diseases: Introducing the D(2) concept. Annu Rev Biophys 37, 215–246 (2008).
de Lau, L. M. & Breteler, M. M. Epidemiology of Parkinson’s disease. Lancet Neurol 5, 525–535 (2006).
Cooper, A. A. et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328 (2006).
Burre, J., Sharma, M. & Sudhof, T. C. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci USA 111, E4274–E4283 (2014).
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q. & Lee, V. M. Synucleins are developmentally expressed and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20, 3214–3220 (2000).
Wang, L. et al. alpha-Synuclein Multimers Cluster Synaptic Vesicles and Attenuate Recycling. Curr Biol 24, 2319–2326 (2014).
Jiang, Z., de Messieres, M. & Lee, J. C. Membrane remodeling by alpha-synuclein and effects on amyloid formation. J Am Chem Soc 135, 15970–15973 (2013).
Lamberto, G. R. et al. Toward the Discovery of Effective Polycyclic Inhibitors of alpha-Synuclein Amyloid Assembly. J Biol Chem 286, 32036–32044 (2011).
Meng, X. Y., Munishkina, L. A., Fink, A. L. & Uversky, V. N. Molecular Mechanisms Underlying the Flavonoid-Induced Inhibition of alpha-Synuclein Fibrillation. Biochemistry-Us 48, 8206–8224 (2009).
Rao, J. N., Dua, V. & Ulmer, T. S. Characterization of alpha-synuclein interactions with selected aggregation-inhibiting small molecules. Biochemistry-Us 47, 4651–4656 (2008).
Lendel, C. et al. On the Mechanism of Nonspecific Inhibitors of Protein Aggregation: Dissecting the Interactions of alpha-Synuclein with Congo Red and Lacmoid. Biochemistry-Us 48, 8322–8334 (2009).
Horvath, I. et al. Mechanisms of protein oligomerization: inhibitor of functional amyloids templates alpha-synuclein fibrillation. J Am Chem Soc 134, 3439–3444 (2012).
Jones, D. R., Moussaud, S. & McLean, P. Targeting heat shock proteins to modulate alpha-synuclein toxicity. Ther Adv Neurol Disord 7, 33–51 (2014).
Breydo, L., Wu, J. W. & Uversky, V. N. alpha-Synuclein misfolding and Parkinson’s disease. Bba-Mol Basis Dis 1822, 261–285 (2012).
Uversky, V. N. et al. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem 277, 11970–11978 (2002).
Park, J. Y. & Lansbury, P. T., Jr. Beta-synuclein inhibits formation of alpha-synuclein protofibrils: a possible therapeutic strategy against Parkinson’s disease. Biochemistry-Us 42, 3696–3700 (2003).
Tsigelny, I. F. et al. Dynamics of alpha-synuclein aggregation and inhibition of pore-like oligomer development by beta-synuclein. Febs J 274, 1862–1877 (2007).
Hashimoto, M. et al. Beta-synuclein regulates Akt activity in neuronal cells. A possible mechanism for neuroprotection in Parkinson’s disease. J Biol Chem 279, 23622–23629 (2004).
da Costa, C. A., Masliah, E. & Checler, F. Beta-synuclein displays an antiapoptotic p53-dependent phenotype and protects neurons from 6-hydroxydopamine-induced caspase 3 activation: cross-talk with alpha-synuclein and implication for Parkinson’s disease. J Biol Chem 278, 37330–37335 (2003).
Lee, D., Paik, S. R. & Choi, K. Y. Beta-synuclein exhibits chaperone activity more efficiently than alpha-synuclein. Febs Lett 576, 256–260 (2004).
Shaltiel-Karyo, R. et al. Inhibiting alpha-synuclein oligomerization by stable cell-penetrating beta-synuclein fragments recovers phenotype of Parkinson’s disease model flies. Plos One 5, e13863 (2010).
Beyer, K., Ispierto, L., Latorre, P., Tolosa, E. & Ariza, A. Alpha- and beta-synuclein expression in Parkinson disease with and without dementia. J Neurol Sci 310, 112–117 (2011).
Rockenstein, E. et al. Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res 914, 48–56 (2001).
Hashimoto, M., Rockenstein, E., Mante, M., Mallory, M. & Masliah, E. beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron 32, 213–223 (2001).
Hashimoto, M. et al. An antiaggregation gene therapy strategy for Lewy body disease utilizing beta-synuclein lentivirus in a transgenic model. Gene Ther 11, 1713–1723 (2004).
Fan, Y. et al. Beta-synuclein modulates alpha-synuclein neurotoxicity by reducing alpha-synuclein protein expression. Hum Mol Genet 15, 3002–3011 (2006).
Rivers, R. C. et al. Molecular determinants of the aggregation behavior of alpha- and beta-synuclein. Protein Sci 17, 887–898 (2008).
Zibaee, S. et al. Sequence determinants for amyloid fibrillogenesis of human alpha-synuclein. J Mol Biol 374, 454–464 (2007).
Roodveldt, C. et al. A Rationally Designed Six-Residue Swap Generates Comparability in the Aggregation Behavior of alpha-Synuclein and beta-Synuclein. Biochemistry-Us 51, 8771–8778 (2012).
Taschenberger, G. et al. Bs-synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Ann Neurol 74, 109–118 (2013).
Galvin, J. E., Uryu, K., Lee, V. M. & Trojanowski, J. Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta- and gamma-synuclein. Proc Natl Acad Sci USA 96, 13450–13455 (1999).
Clore, G. M. & Iwahara, J. Theory, practice and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem Rev 109, 4108–4139 (2009).
Tang, C., Ghirlando, R. & Clore, G. M. Visualization of transient ultra-weak protein self-association in solution using paramagnetic relaxation enhancement. J Am Chem Soc 130, 4048–4056 (2008).
Wu, K. P. & Baum, J. Detection of Transient Interchain Interactions in the Intrinsically Disordered Protein alpha-Synuclein by NMR Paramagnetic Relaxation Enhancement. J Am Chem Soc 132, 5546; PMCID: PMC3064441 (2010).
Fawzi, N. L., Doucleff, M., Suh, J. Y. & Clore, G. M. Mechanistic details of a protein-protein association pathway revealed by paramagnetic relaxation enhancement titration measurements. P Natl Acad Sci USA 107, 1379–1384 (2010).
Clayton, D. F. & George, J. M. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci 21, 249–254 (1998).
Polevoda, B. & Sherman, F. N-terminal Acetyltransferases and Sequence Requirements for N-terminal Acetylation of Eukaryotic Proteins. J Mol Biol 325, 595–622 (2003).
Sung, Y. H. & Eliezer, D. Residual structure, backbone dynamics and interactions within the synuclein family. J Mol Biol 372, 689–707 (2007).
Bertoncini, C. W. et al. Structural characterization of the intrinsically unfolded protein beta-synuclein, a natural negative regulator of alpha-synuclein aggregation. J Mol Biol 372, 708–722 (2007).
Bourhim, M., Kruzel, M., Srikrishnan, T. & Nicotera, T. Linear quantitation of Abeta aggregation using Thioflavin T: reduction in fibril formation by colostrinin. J Neurosci Methods 160, 264–268 (2007).
Groenning, M. et al. Binding mode of Thioflavin T in insulin amyloid fibrils. J Struct Biol 159, 483–497 (2007).
Peterson, D. W., Zhou, H. J., Dahlquist, F. W. & Lew, J. A soluble oligomer of tau associated with fiber formation analyzed by NMR. Biochemistry-Us 47, 7393–7404 (2008).
Kang, L. J. et al. N-terminal acetylation of alpha-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci 21, 911–917; PMCID; PMC3403430 (2012).
Fitzkee, N. C., Masse, J. E., Shen, Y., Davies, D. R. & Bax, A. Solution Conformation and Dynamics of the HIV-1 Integrase Core Domain. J Biol Chem 285, 18072–18084 (2010).
Vilar, M. et al. The fold of alpha-synuclein fibrils. P Natl Acad Sci USA 105, 8637–8642 (2008).
Comellas, G. et al. Structured Regions of alpha-Synuclein Fibrils Include the Early-Onset Parkinson’s Disease Mutation Sites. J Mol Biol 411, 881–895 (2011).
Karamanos, T. K., Kalverda, A. P., Thompson, G. S. & Radford, S. E. Visualization of Transient Protein-Protein Interactions that Promote or Inhibit Amyloid Assembly. Mol Cell 55, 214–226 (2014).
Marsh, J. A., Teichmann, S. A. & Forman-Kay, J. D. Probing the diverse landscape of protein flexibility and binding. Curr Opin Struc Biol 22, 643–650 (2012).
Lamberto, G. R. et al. Structural and mechanistic basis behind the inhibitory interaction of PcTS on alpha-synuclein amyloid fibril formation. P Natl Acad Sci USA 106, 21057–21062 (2009).
Mirecka, E. A. et al. Sequestration of a beta-Hairpin for Control of alpha-Synuclein Aggregation. Angew Chem Int Edit 53, 4227–4230 (2014).
Fonseca-Ornelas, L. et al. Small molecule-mediated stabilization of vesicle-associated helical alpha-synuclein inhibits pathogenic misfolding and aggregation. Nat Commun 5, 5857–5868 (2014).
Guo, J. L. & Lee, V. M. Y. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20, 130–138 (2014).
Clinton, L. K., Blurton-Jones, M., Myczek, K., Trojanowski, J. Q. & LaFerla, F. M. Synergistic Interactions between A beta, Tau and alpha-Synuclein: Acceleration of Neuropathology and Cognitive Decline. J Neurosci 30, 7281–7289 (2010).
Irwin, D. J., Lee, V. M. Y. & Trojanowski, J. Q. Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 14, 626–636 (2013).
Kurosinski, P., Guggisberg, M. & Gotz, J. Alzheimer’s and Parkinson’s disease-overlapping or synergistic pathologies? Trends Mol Med 8, 3–5 (2002).
Kang, L. J., Wu, K. P., Vendruscolo, M. & Baum, J. The A53T Mutation is Key in Defining the Differences in the Aggregation Kinetics of Human and Mouse alpha-Synuclein. J Am Chem Soc 133, 13465–13470 (2011).
Delaglio, F. et al. Nmrpipe-a Multidimensional Spectral Processing System Based on Unix Pipes. J Biomol Nmr 6, 277–293 (1995).
Goddard, T. D. & Kneller, D. G. SPARKY 3.
Elzhov, T. M. & Katharine, M. Spiess Andrej-Nikolai; Bolker, B. minpack.lm: R interface to the Levenberg-Marquardt nonlinear least-squares algorithm found in MINPACK, plus support for bounds <https://cran.r-project.org/web/packages/minpack.lm/minpack.lm.pdf>, (2013) (Date of access:09/04/2015).
Acknowledgements
We would like to thank Gina Moriarty and Dr. Ana Monica Nunes for helpful and stimulating discussions and Dr. Valentin Starovoytov for assistance with TEM experiments. This work was supported by grant GM110577 from the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
M.J. designed, performed, analyzed experiments and wrote the manuscript, K.-P.W. developed the methodology for NMR PRE titration experiments, J.B. designed and supervised experiments and data analysis and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Janowska, M., Wu, KP. & Baum, J. Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein. Sci Rep 5, 15164 (2015). https://doi.org/10.1038/srep15164
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15164
This article is cited by
-
Transcriptional mutagenesis of α-synuclein caused by DNA oxidation in Parkinson’s disease pathogenesis
Acta Neuropathologica (2023)
-
Increased Dynamics of α-Synuclein Fibrils by β-Synuclein Leads to Reduced Seeding and Cytotoxicity
Scientific Reports (2019)
-
Investigating the neuroprotective effect of AAV-mediated β-synuclein overexpression in a transgenic model of synucleinopathy
Scientific Reports (2018)
-
The increasing dynamic, functional complexity of bio-interface materials
Nature Reviews Chemistry (2018)
-
β-Synuclein suppresses both the initiation and amplification steps of α-synuclein aggregation via competitive binding to surfaces
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
