
Abstract
Fluorescent tracking gene delivery could provide us with a better understanding of the critical steps in the transfection process. However, for in vivo tracking applications, a small diameter (<10 nm) is one of the rigorous requirements for tracking vectors. Herein, we have demonstrated a new paradigm for two-photon tracking gene delivery based on a dendritic nano-sized hexanuclear ruthenium(II) polypyridyl complex. Because this metallodendrimer has a multivalent periphery, the complex, which is 6.1 nm, showed high stability and excellent dispersibility and could stepwise condense DNA in vitro. With the outstanding photochemical properties of Ru(II) polypyridyl, this complex could track gene delivery in vivo using one- and two-photon imaging.
Similar content being viewed by others

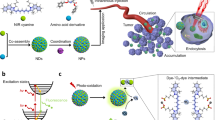

Introduction
Gene therapy is a promising technique for treating human disease by the transfer of genetic material into specific cells within the patient1,2,3. Compared with viral vectors in vivo, nonviral gene delivery systems have gained immense importance in terms of safety4,5,6,7,8. As nonviral gene vectors, nanomaterials have been widely used to deliver genetic material in vivo9,10,11,12,13 and some of them can function as fluorescent tracking vectors14,15,16,17,18,19,20, which could provide us with a better understanding of the critical steps in the transfection process21. The tracking nanomaterials are usually labelled with organic dyes14,15, quantum dots16,17,18, carbon dots19 or metal complexes20. To apply these materials for tracking in vivo specifically, the small diameter (<10 nm) is one of the rigorous requirements for nanomaterials22,23,24,25. However, synthesizing small tracking nanomaterials generally requires multiple complicated reactions. Additionally, quantum dot-based nanomaterials contain toxic heavy metals, while small organic dyes suffer from small Stokes shifts and rapid photobleaching26,27,28. Even worse, the introduction of fluorescent materials may alter the delivery mechanisms and increase potential side-effects29.
Very recently, several efforts have been devoted to the development of multinuclear complexes that induce DNA condensation due to their greater variety of charge states and can be used as non-viral gene vectors21,30,31,32,33,34. As multinuclear complexes with metal fragments located within the dendrimers or at the periphery, metallodendrimers have applications in sensing, catalysis, biomedicine and materials science35,36,37,38,39,40,41 based on specific properties, including highly controlled nanosize, tunable solubility and multivalent periphery42. However, using a Ru(II) polypyridyl metallodendrimer-based vector to track intracellular gene delivery has been rarely explored. Moreover, Ru(II) polypyridyl complexes have outstanding photochemical properties, such as high photostability, large Stokes shifts, great one- and two-photon luminescence properties and large two-photon absorption cross sections, which have made these complexes promising luminescent tools for two-photon tracking studies43,44,45,46. Therefore, we attempted to employ a dendritic luminophore with proton buffering capacity polybenzimidazole ligands30,31 as a tracking non-viral gene vector. This complex would allow us to observe gene condensing in vitro and track gene delivering in vivo.
Herein, a dendritic hexanuclear Ru(II) polypyridyl complex based on polybenzimidazole ligands, Ru6L (Fig. 1), was synthesized and developed as a one- and two-photon luminescent tracking non-viral gene vector. The photophysical properties and the size of Ru6L were characterized. After studying the interactions of Ru6L with DNA and the DNA condensation mechanism in vitro, the cellular uptake of both the complex and the Ru6L-DNA particles was investigated. Then, the complex was used as a one- and two-photon luminescent tracking non-viral gene vector. Finally, the transfection efficiency and cytotoxicity of the DNA condensates were evaluated by luciferase assays and MTT assays.

The synthetic route of complex Ru6L.
Results and Discussion
The synthesis of the ligand and complex was outlined in Fig. 1. The bridging ligand 1,2,3,4,5,6-hexakis{4-((1,10-phenanthroline-[5,6-d]imidazol-2-yl)-phenoxy)methyl}benzene (L) was obtained at a 58% yield through condensation of 1,10-phenanthroline-5,6-dione with 1,2,3,4,5,6-hexakis[(4-formylphenoxy)methyl]benzene in refluxing glacial acetic acid. The 1H NMR spectrum of ligand L was not obtained due to its poor solubility in common NMR solvents. Complex {[Ru(bpy)2]6L}(ClO4)12 (Ru6L) was prepared by refluxing Ru(bpy)2Cl2.2H2O and L in ethylene glycol and isolated as a ClO4– salt, with a 62% yield. This complex was characterized by 1H NMR spectroscopy, IR spectroscopy, ESI-MS and elemental analysis (Figure S1-S3).
The photophysical data of Ru6L are summarized in Table S1. The absorption band at approximately 400–550 nm was assigned to metal-ligand charge transfer (MLCT) absorption (Figure S4). The maximum emission wavelength (λem) was 612 nm, with a lifetime of 110 ns. The emission quantum yield was 0.027 and the largest two-photon absorption (TPA) cross-section was 175 GM at 830 nm with reference to rhodamine B (Figure S5). The complex was a perfectly monodisperse nanoparticle with 12 positive charges derived from Ru2+ ions (Figure S6). The diameter of Ru6L was determined to be 6.1 nm by TEM (Fig. 2), which was similar to the hydrodynamic diameter 6.7 ± 0.3 nm determined by DLS (Figure S6).

A TEM image of complex Ru6L obtained on a carbon-coated Cu grid.
We investigated the complex’s ability to condense DNA. The first study examined the migration of Ru6L-pBR 322 DNA by gel retardation assays with a load of 45 μM (bp) DNA in each well. As shown in Fig. 3, with increasing the concentrations of Ru6L (0-6 μM), pBR 322 DNA gradually diminished and was retained in the well at a +/− ratio (total positive charge/total negative charge) of 1.1. The primary DNA condensation-driving forces were considered electrostatic interactions by the multivalent periphery of Ru6L. The DNA binding ability was determined by a DNA titration approach with CT-DNA (calf thymus DNA). The binding constant was calculated to be 1.75 × 105 M−1 (Figure S7).

(a) Agarose gel electrophoresis of pBR 322 DNA (45 μM) after incubation with Ru6L at various +/− ratios in aqueous solution; (b) Hydrodynamic diameter and (c) zeta potential of the pBR 322 DNA (1.5 μM) incubation with Ru6L at various +/− ratios in an aqueous solution,as determined by DLS.
The average hydrodynamic diameter of Ru6L-pBR 322 DNA particles at various +/− ratios in aqueous solution is shown in Fig. 3. The particle diameter was stable at approximately 140 nm when the +/− ratio was 4 or higher, while the zeta potential increased as the +/− ratio increased. To visually investigate the condensation process, we performed AFM of the Ru6L-2kb DNA particles at various +/− ratios (Fig. 4). When the +/− ratio was 1, local DNA bending, long-range cross-links and micro-loops were observed. Upon increasing the +/− ratio to 2, small particles were observed at the middle or the tail of the DNA duplex. As the +/− ratio increased, highly condensed DNA molecules were observed and they condensed into compact hemispherical structures at a +/− ratio of 20. Ru6L-pBR 322 DNA particles at a +/− ratio of 20 were also observed as well-distributed DNA particles with a diameter from 50 to 90 nm (Figure S8).

(a) AFM images of the condensation process, starting with2 kb DNA (1.5 μM), induced by incubation with Ru6L at +/− ratios of 1 (a1), 2 (a2), 4 (a3) and 20 (a4), respectively. (b) Schematic representation of the stepwise DNA compaction by Ru6L.
In addition, we performed a DNA nuclease-catalyzed biodegradation assay (Figure S9a), a photocleavage assay (Figure S9b) and a continuous irradiation assay (Figure S10) with Ru6L. The results showed that Ru6L displayed no photocleavage or photoreactivity toward DNA; in contrast, it protected DNA against nuclease-catalyzed biodegradation.
Before performing cellular uptake studies of Ru6L-DNA particles, we investigated the mechanism of cellular uptake of Ru6L using confocal microscopy (Figure S11). Generally speaking, the cellular uptake of small molecules can occur through energy-independent (facilitated diffusion, passive diffusion) and energy-dependent (endocytosis, active transport) pathways. To determine whether Ru6L entered the cell via an energy-independent or energy-dependent transport pathway, HeLa cells were either incubated with Ru6L at 4 °C or pretreated with the metabolic inhibitors 2-deoxy-D-glucose and oligomycin. Figure S11b and S11c show that cellular luminescence was significantly suppressed in cases when the cells were incubated with the complex at 4 °C or pretreated with the metabolic inhibitors, indicating that Ru6L uptake followed an energy-dependent pathway. Endocytosis is well known as the most common energy-dependent pathway by which eukaryotic cells uptake extracellular materials. Endocytosis is also affected by temperature or adenosine triphosphate (ATP). We used the endocytic inhibitors chloroquine and NH4Cl to examine the role of this pathway in Ru6L uptake. As observed from the relative intensities and location of Ru6L in the cells after treatment with these inhibitors (Figure S11d and S11e), they had no effect on the cellular uptake of Ru6L, which indicates that Ru6L is not taken up by living cells via an endocytosis pathway. This finding is not surprising because cellular uptake mechanisms are generally complex and diverse.
Flow cytometry was used to quantify the intracellular uptake of the DNA particles at the +/− ratio of 20 (Figure S12). The result indicated that most of cells took up the DNA particles after a 4 h incubation in serum-free DMEM. We examined the localization of DNA particles in HeLa cells by TEM. TEM micrographs confirmed the uptake of the particles by endocytosis and showed that the DNA particles made HeLa cell form an endosome after they entered the HeLa cells (Fig. 5). Particles were also found in the cytoplasm, meaning that the particles successfully escaped from endosomes, which was important for transfection. The particle size was approximately 50 nm, as measured by AFM. The mechanism of cellular uptake of the DNA particles was also studied (Figure S13). The cellular luminescence of Figure S13b which incubated with the particles at 4 °C was significantly suppressed, it means the particles entered the cell via an energy-independent transport pathway. After treatment with the metabolic inhibitors (Figure S13c) and the endocytic inhibitors (Figure S13d and S13e), no effect was found in the cells which treated with the metabolic inhibitors, but the cellular luminescence which treated with the endocytic inhibitors was suppressed, the results indicated that Ru6L-pEGFP DNA particles were taken up by living cells via an endocytosis pathway.

Cellular uptake and intracellular localization of Ru6L-pEGFP DNA particles at the +/− ratio of 20 monitored, as by TEM. The DNA concentration was 1.5 μM, N: nucleus, C: cytoplasm.
To investigate the intracellular behaviors of Ru6L-pEGFP DNA particles, one- and two-photon fluorescence microscopy was used to monitor the time-dependent transport and transfection of pEGFP DNA plasmids that were condensed by Ru6L (Fig. 6). We stained the nuclei of HeLa cells with Hoechst 33258; then, the DNA particles were added to the cells. In 20 min, the Ru6L-DNA particles attached to the cell membrane quickly due to their high positive zeta potential. After 4 h, particles were found in the cytoplasm. Then, the culture medium was replaced with fresh DMEM containing 10% fetal bovine serum (FBS). After another 12 h, EGFP expression was detected and higher EGFP expression was found after 24 h. Similar behaviors were observed under one-photon and two-photon excitation. In addition, due to the two-photon absorption, the background signal was strongly suppressed and a higher resolution image was obtained using two-photon confocal laser scanning microscopy.

Time-dependent confocal microscopy images of entry and transportation of Ru6L-pEGFP DNA particles at the +/− ratio of 20 in HeLa cells. The DNA concentration was 1.5 μM. The red luminescence is Ru6L, the blue florescence is Hoechst 33258 and the green florescence is GFP.
The most commonly used method for intracellular plasmid trafficking is the fluorescent labeling of non-viral vectors with organic dyes14,15. It should be pointed out that to track the dynamic changes during a specific period of time, the dye must possess improved photostability and must be photostable under continual irradiation with light from fluorescent microscopes. However, most organic dyes have notable shortcomings, including poor water solubility, high toxicity to living cells and poor photostability. Organic dyes may also cause extensive cellular damage and unwanted background signal due the ultraviolet (UV) radiation required for their excitation and small Stokes shifts26,27. The short excitation wavelengths (<650 nm) also inhibit the use of these materials in thick tissues or live animals due to the resultant short penetration depth47. In addition, the introduction of dyes may alter the delivery mechanisms and have increased side-effects29. The use of Ru6L with intrinsic two-photon luminescence as DNA carriers is an attractive solution to these problems because this complex exhibits near infrared (NIR) excitation wavelengths48.
We determined the relative transfection efficiency of this nonviral system by luciferase assays and plasmid pGL3 was used a control vector (Fig. 7). As a control, applying DNA only resulted in a low level of luciferase expression. The luciferase expression was increased when Ru6L was used. The luciferase expression was stable when the +/− ratios of the particles ranged from 12 to 24 and the highest luciferase expression was in response to the +/− ratio of 20.

Transfection efficiencies of Ru6L-pGL3 DNA particles in HeLa cells, as determined by luciferase assays; the DNA concentration was 1.5 μM. As a control, DNAand lipofectamine 2000 were also investigated.
Cytotoxicity of non-viral gene vectors is one of the major concerns of gene delivery. Therefore, we examined the viability of HeLa cells treated with the complex and Ru6L-pEGFP DNA particles (Fig. 8). With increasing concentrations, the viability of the HeLa cells decreased slowly for both the complex and the particles. Although Ru6L exhibited the related cytotoxicity as the concentration increased, the viability of HeLa cells was more than 80% at a concentration of 5 μM. The results indicated that this transfection system had relatively low cytotoxicity.

The cell viability of Ru6L and Ru6L-pEGFP DNA particles in HeLa cells, as determined by MTT assays; the DNA concentration was 1.5 μM.
Conclusion
In the present study, we have designed and synthesized a dendritic nano-sized hexanuclear Ru(II) polypyridyl complex (Ru6L) based on polybenzimidazole ligands. This complex has a multivalent periphery and a nanosize of 6.1 nm. Gel retardation assay, TEM, AFM and dynamic light scattering studies show that Ru6L exhibits high stability and excellent dispersibility and could stepwise condense DNA in vitro. More interestingly, Ru6L was applied as a non-viral gene vector for tracking DNA delivery in live cells using one- and two-photon fluorescence microscopes. With two-photon microscopy, a high signal-to-noise contrast was achieved by irradiation with an 830 nm laser. Our work provides new insights into improving real-time tracking during gene delivery and transfection as well as important information for the design of multifunctional non-viral vectors.
Methods
All reagents were purchased from commercial sources and used without further purification unless otherwise specified. The plasmid pBR 322 DNA was obtained from MBI Fermentas, the plasmid pEGFP DNA was purchased from Clontech and the plasmid pGL3 control vector and luciferase kid were obtained from Promega. Unless otherwise stated, the DNA concentrations are expressed in base pairs. All samples were prepared using distilled water that had been passed through a Millipore-Q ultra-purification system. The compounds 1,10-phenanthroline-5,6-dione49 and [Ru(bpy)2]Cl2.2H2O50 were prepared according to literature methods. The complex was dissolved in DMSO prior to the experiments. Then, the calculated quantities of the complex solutions were added to the appropriate medium to yield a final DMSO concentration of less than 1% (v/v).
Microanalyses (C, H and N) were performed with a vario EL cube elemental analyzer. Infrared spectra were obtained with a Nicolet 170SX-FTIR spectrophotometer and KBr discs. Electrospray ionization mass spectra (ESI-MS) were recorded on an LCQ system (Finnigan MAT, USA).Fast atom bombardment mass spectrometry (FAB-MS) was recorded using a VG ZAB-HS. 1H NMR spectra were recorded on a Varian-500 spectrometer at 25 °C. All chemical shifts are given relative to tetramethylsilane (TMS). The UV-Vis spectra were recorded on a Perkin-Elmer Lambda 850 spectrophotometer. Emission spectra were recorded on a Perkin-Elmer LS 55 spectrophotometer at room temperature (25 °C). Time-resolved emission measurements were conducted on an FLS 920 combined fluorescence-lifetime and steady-state spectrometer. Quantum yields of luminescence at room temperature (25 °C) were calculated according to literature procedures using [Ru(bpy)3]2+ (φ = 0.028 in aerated aqueous solution) as the reference emitter51. All date were processed using the Origin 8 software package.
Atomic force microscopy (AFM) images were obtained in air at room temperature with an SPA400 atomic force microscope unit and an SPI3800N control station (Seiko Instruments) operated in the tapping mode. Probes were made of a single silicon crystal with a cantilever length of 129 mm and a spring constant of 33–62N/m (OMCLAC160TS-W2, Olympus). Dynamic light scattering and zeta potential experiments were performed using dynamic laser light scattering equipment (DLS, Brooken Haven BI-200SM). Transmission electron micrographs were obtained with a JEM100CX electron microscope.
Synthesis of 1,2,3,4,5,6-hexakis[(4-formylphenoxy)methyl]benzene (Q)
A mixture of 4-hydroxybenzaldehyde (0.50 g, 4.09 mmol), 1,2,3,4,5,6-hexakis(bromomethyl)benzene (0.30 g, 0.47 mmol) and K2CO3 (0.60 g, 4.35 mmol) in DMF (30 mL) was heated to 80 °C for 24 h under nitrogen. After cooling to room temperature, the solution was diluted with 300 mL of water. The mixture was filtered and the solid was washed successively with boiling MeOH, CHCl3 and Et2O, affording the desired product as a white solid. Yield: 0.21 g (50%). Anal. Calcd. for C54H42O12: C, 73.46; H, 4.79. Found: C, 73.24; H, 4.80. IR νmax (KBr, cm−1): 1689 (CHO).1H NMR (500 MHz, d6-DMSO) δ 9.76 (s, 6H), 7.70 (d, J = 15 Hz, 12H), 7.04 (d, J = 15 Hz, 12H), 5.42 (s, 12H). ESI-MS: m/z = 905.4 [M+Na]+.
Synthesis of 1,2,3,4,5,6-hexakis{4-((1,10-phenanthroline-[5,6-d]imidazol-2-yl)- phenoxy)methyl}benzene (L)
A mixture of Q(0.15 g, 0.17 mmol), 1,10-phenanthroline-5,6-dione (0.30 g, 1.42 mmol) and NH4OAc (2.00 g, 25.64 mmol) in glacial acetic acid (20 mL) was heated to 130 °C for 8 h, yielding a suspension. The reaction mixture was filtered while hot and the solid was washed successively with EtOH, hot DMF and Et2O, affording the desired product as a yellow solid. Yield: 0.20 g (58%). Anal. Calcd. for C126H78N24O6: C, 74.77; H, 3.88; N, 16.61. Found: C, 74.55; H, 4.16; N, 16.43. IR νmax (KBr, cm−1): 3392 (N-H). FAB-MS: m/z = 2025 [M+1]. The 1H NMR spectrum was not obtained due to its poor solubility in common NMR solvents.
Synthesis of complex {[Ru(bpy)2]6L}(ClO4)12 (Ru6L)
A mixture of L (0.075 g, 0.037 mmol) and Ru(bpy)2Cl2.2H2O (0.15 g, 0.29 mmol) in ethylene glycol (10 mL) was heated to 180 °C for 14 h under nitrogen to give a clear deep-red solution. The cooled reaction mixture was diluted with water (15 mL). Saturated aqueous sodium perchlorate solution was added under vigorous stirring and the solution filtered. The dark red solid was collected, washed with small amounts of water and diethyl ether, dried under vacuum and purified by column chromatography on alumina using acetonitrile-ethanol as the elutant. The solvent was removed under reduced pressure and red microcrystals were obtained. Yield: 0.13 g (62%). Anal. Calcd. for C246H174Cl12N48O54Ru6: C, 51.85; H, 3.08; N, 11.80. Found: C, 51.64; H, 3.37; N, 11.57.IR νmax (KBr, cm−1): 3360 (N-H) and 1087 (ClO4−). ESI-MS: m/z = 423.6 [M-6ClO4-6H]12+, 438.8 [M-9ClO4-6H]11+, 453.8 [M-7ClO4-6H]11+, 481.9 [M-9ClO4-6H]10+, 500.8 [M-12ClO4+4H]9+, 512.0 [M-11ClO4-5H]9+, 554.5 [M-7ClO4-6H]9+, 574.9 [M-11ClO4-4H]8+, 600.5 [M-9ClO4+2H]8+, 642.7 [M-12ClO4-5H]7+, 657.6 [M-11ClO4]7+, 672.4 [M-10ClO4-3H]7+, 750.5 [M-12ClO4-H]6+, 766.5 [M-11ClO4-4H]6+, 782.8 [M-10ClO4-6H]6+, 799.4 [M-9ClO4-6H]6+, 815.5 [M-8ClO4-6H]6+, 832.8 [M-7ClO4-6H]6+, 901.3 [M-12ClO4+2H]5+, 921.0 [M-11ClO4-H]5+, 940.5 [M-10ClO4]5+, 959.0 [M-9ClO4-6H]5+, 979.7 [M-8ClO4-3H]5+. 1H NMR (500 MHz, d6-DMSO) δ 9.10 (bs, 12H), 8.81 (q, J = 5 Hz, 24H), 8.21 (d, J = 10 Hz, 12H), 8.20 (t, J = 7.5 Hz, 12H), 8.04 (t, J = 7.5 Hz, 12H), 7.95 (s, 12H), 7.82 (d, J = 5 Hz, 24H), 7.54–7.59 (m, 24H), 7.28 (q, J = 5 Hz, 24H), 5.56 (bs, 12H).
Determining the size of the complex
The complex Ru6L was dissolved in CH3CN at a concentration of 5 μM. The solution was drop cast onto a carbon-coated Cu grid and the excess solution was absorbed by filter paper to avoid aggregation. The TEM image of the drop cast sample was taken with a JEM100CX electron microscope.
Dynamic laser light scattering equipment was used to determine the average hydrodynamic diameter and the zeta potential of Ru6L at a concentration of 5 μM in distilled water that had been passed through a Millipore-Q ultra-purification system. Typically, 5 runs were measured for the solution and the average of all runs is reported.
Determination of two-photon absorption cross-sections
The two-photon absorption (TPA) spectra of complex were determined over a broad spectral region by a two-photon induced luminescence (TPL) method relative to Rhodamine B in methanol as the standard. The two-photon luminescence data were acquired using an OpoletteTM 355II (pulse width ≤ 100 fs, 80 MHz repetition rate, tuning range 730–890 nm, Spectra Physics, Inc., USA). Two-photon luminescence measurements were performed in fluorometric quartz cuvettes. The experimental luminescence excitation and detection conditions were conducted with negligible reabsorption processes, which can affect TPA measurements. The quadratic dependence of the two-photon induced luminescence intensity on the excitation power was verified at an excitation wavelength of 830 nm. The two-photon absorption cross-section of the complex was calculated at each wavelength according to Equation (1)52:

where I is the integrated luminescence intensity, C is the concentration, n is the refractive index and φ is the quantum yield. The subscript ‘1’ refers to the reference samples and ‘2’ for the experimental samples.
Preparation of DNA particles
The DNA particles were prepared by incubating the mixtures containing DNA and Ru6L at the given +/− ratios in 50 mM Tris-HCl (Tris = Tris(hydroxymethyl) aminomethane) solution (pH = 7.4) or in cell culture, followed by vortexing for 30 min to allow equilibration at room temperature.
Gel retardation assay
Negative supercoiled pBR 322 DNA was treated with Ru6L in 50 mM Tris-HCl solution (pH = 7.4) and the solutions were analyzed by electrophoresis for 1.5 h at 75 V on a 1% agarose gel in TBE buffer (89 mM Tris-borate acid, 2 mM EDTA, pH = 8.3). The gel was stained with 1 μg/mL ethidium bromide (EB) and photographed on an Alpha Innotech IS-5500 fluorescence chemiluminescence and visible imaging system.
Dynamic light scattering and zeta potential assay
Dynamic laser light scattering equipment was used to determine the average hydrodynamic diameter and the zeta potential of Ru6L-pBR 322 DNA particles at various +/− ratios in 50 mM Tris-HCl solution (pH = 7.4). Typically, 5 runs were measured for each solution, with the average of all the runs reported.
AFM imaging
The 2kb DNA fragment was generated by PCR using pBR 322 plasmid DNA as a template53. The condensation process of Ru6L-2kb DNA and the morphologies Ru6L-pBR 322 DNA were examined with AFM. Samples were dropwise-added (10 μL) onto a mica substrate, which was freshly cleaved by pulling off the top sheets with tape. One min later, the substrate was spin coated (1400 rpm, 30 s) and rinsed with 20 μL of distilled water. AFM images were obtained in air at room temperature. The images were captured in a 256 × 256 pixels format and analyzed with the software accompanying the imaging module.
Cell culture and cell viability assay
HeLa cells were maintained as monolayer cultures in DMEM supplemented with 10% fetal bovine serum (FBS) at 37 °C under 5% CO2. Exponentially grown HeLa cells were seeded in triplicate onto 96-well plates at 1 × 104 cells/well. After incubation for 24 h, the cells were treated with increasing concentrations of the tested complex and Ru6L-pEGFP DNA particles for 24 h. To stain the viable cells, 20 μL of MTT (5 mg/mL) was added to each well. The cells were then incubated for 4 h at 37 °C. After the medium was carefully aspirated without disturbing the formed crystals, the dye was dissolved in 200 μL of DMSO. A Tecan Infinite M200 monochromator-based multifunction microplate reader was used to measure the optical density of each well, with background subtraction at 590 nm. The cell survival rate in the control wells without Ru6L solutions were considered as 100% cell survival.
The mechanism of cellular uptake of Ru6L
The cells were trypsinized, counted and adjusted to 1 × 105 cells mL−1 and 1 mL was added to five 35-mm2 Petri dishes (MatTek, USA) for laser confocal microscopy. After 24 h, the first dish was incubated with 5 μM Ru6L at 37 °C for 2 h. The second dish was incubated with 5 μM Ru6L at 4 °C for 2 h. The third dish was pretreated with 50 mM 2-deoxy-D-glucose and 5 μM oligomycin in PBS for 1 h at 37 °C and then incubated with 5 μM Ru6L at 37 °C for 2 h. The fourth and the fifth dishes were pretreated with endocytic inhibitors NH4Cl (50 mM) and chloroquine (50 μM) for 30 min respectively and then incubated with 5 μM Ru6L at 37 °C for 2 h.
After being washed with fresh PBS (pH = 7.0) three times, the cells were imaged on a Zeiss LSM 710 NLO confocal microscope (63 × /NA 1.4 oil immersion objective). For two-photon images, the excitation wavelength of the laser was 830 nm for Ru6L and the emission spectra were integrated over 580-630 nm (single channel).
Cellular uptake of Ru6L-DNA particles
The cells were trypsinized, counted and adjusted to 1 × 104 cells mL−1 and 1 mL of the cell solution was added to each plate. After 24 h, the cell culture medium was replaced with 800 μL serum-free DMEM. Ru6L-pEGFP DNA particles at the +/− ratio of 20, containing 1 μg pEGFP DNA in 200 μL serum-free DMEM, was added to the cells and the cells were incubated at 37 °C for 4 h.
For flow cytometry, the cells were washed with PBS three times, trypsinized and centrifuged in PBS. Cells were harvested and single cell suspensions in 0.5 mL PBS were prepared and subjected to flow cytometric analysis. A flow cytometer (Coulter Co. USA) was used to measure the fluorescent intensity, with excitation at 488 nm.
For TEM imaging analysis, cell processing was performed in situ, without displacement from the culture dish. Cells were fixed in 0.1 M PBS containing 2.5% glutaraldehyde and 4% paraformaldehyde for 1 h, rinsed with distilled water, stained with 0.5% uranyl acetate for 1 h, dehydrated in a graded series of ethanol (30, 60, 70, 90 and 100%) and embedded in epoxy resin. The resin was polymerized at 60 °C for 48 h. Ultrathin sections (50–75 nm) obtained with an LKB ultramicrotome were stained with 2% aqueous uranyl acetate and 2% aqueous lead citrate.
The mechanism of cellular uptake of Ru6L-DNA particles
The cells were trypsinized, counted and adjusted to 1 × 105 cells mL−1 and 1 mL was added to five 35-mm2 Petri dishes (MatTek, USA) for laser confocal microscopy. After 24 h, the first dish was incubated with Ru6L-pEGFP DNA particles at the +/− ratio of 20 at 37 °C for 4 h. The second dish was incubated with Ru6L-pEGFP DNA particles at the +/− ratio of 20 at 4 °C for 4 h. The third dish was pretreated with 50 mM 2-deoxy-D-glucose and 5 μM oligomycin in PBS for 1 h at 37 °C and then incubated with Ru6L-pEGFP DNA particles at the +/− ratio of 20 at 37 °C for 4 h. The fourth and the fifth dishes were pretreated with endocytic inhibitors NH4Cl (50 mM) and chloroquine (50 μM) for 30 min respectively and then incubated with Ru6L-pEGFP DNA particles at the +/− ratio of 20 at 37 °C for 4 h.
After being washed with fresh PBS (pH = 7.0) three times, the cells were imaged on a Zeiss LSM 710 NLO confocal microscope (63 × /NA 1.4 oil immersion objective). For two-photon images, the excitation wavelength of the laser was 830 nm for Ru6L and the emission spectra were integrated over 580-630 nm (single channel).
One- and two-photon luminescent imaging
The cells were trypsinized, counted and adjusted to 1 × 105 cells mL−1 and 1 mL of the cell suspension was added to a 35-mm2 Petri dish (MatTek, USA) for laser confocal microscopy. After 24 h, the cell culture medium was replaced with 800 μL serum-free DMEM. Ru6L-pEGFP DNA particles at the +/− ratio of 20, containing 1 μg pEGFP DNA in 200 μL serum-fee DMEM, was added to the cells and the cells were incubated at 37 °C for 4 h. Then, the medium was replaced with fresh DMEM containing 10% FBS and the cells were incubated for various timed (20 min and 4, 12 and 24 h).
After being washed with fresh PBS (pH = 7.0) three times, the cells were imaged on a Zeiss LSM 710 NLO confocal microscope (63 × /NA 1.4 oil immersion objective). The excitation wavelength of the laser was 488 nm and the emission spectra were integrated over 580-630 nm (single channel). For two-photon images, the excitation wavelength of the laser was 830 nm for Ru6L.
Luciferase assay
HeLa cells were seeded onto a 96-well cell-culture plate at a cell density of 1 × 104 cells per well and then incubated for 24 h. Cells were washed with PBS three times and then cultured with serum-free DMEM. Ru6L-pGL3 DNA control vector particles with increasing concentrations of the tested complex containing 0.2 μg plasmid were added to the cells and the cells were incubated at 37 °C for 4 h. The medium was then replaced with fresh DMEM with 10% FBS and the cells were incubated for an additional 24 h. Cells were washed with PBS, harvested and treated for 30 min at 4 °C with end-over-end rotation in lysis buffer (50 mM Tris-HCl, pH = 7.5, 150 mM NaCl, 2% Triton X-100, 2% NP40). The luciferase assay was performed according to the manufacturer’s protocol (Promega). Relative light units (RLUs) were measured with Varioskan Flash (Thermo Scientific, USA) and a GloMaxTM 96 microplate luminometer (Promega, USA).
Additional Information
How to cite this article: Qiu, K. et al. A dendritic nano-sized hexanuclear ruthenium(II) complex as a one- and two-photon luminescent tracking non-viral gene vector. Sci. Rep. 5, 10707; doi: 10.1038/srep10707 (2015).
References
Mulligan, R. C. The basic science of gene therapy. Science 260, 926–932 (1993).
Ferber, D. Gene therapy: safer and virus-free? Science 294, 1638–1642 (2001).
Ibraheem, D., Elaissari, A. & Fessi, H. Gene therapy and DNA delivery systems. Int. J. Pharm. 459, 70–83 (2014).
Luo, D. & Saltzman, W. M. Synthetic DNA delivery systems. Nat. Biotechnol. 18, 33–37 (2000).
Thomas, C. E., Ehrhardt, A. & Kay, M. A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 4, 346–358 (2003).
Niidome, T. & Huang, L. Gene therapy progress and prospects: nonviral vectors. Gene Ther. 9, 1647–1652 (2002).
He, C. X., Tabata, Y. & Gao, J. Q. Non-viral gene delivery carrier and its three-dimensional transfection system. Int. J. Pharm. 386, 232–242 (2010).
Merkel, O. M. & Kissel, T. Nonviral pulmonary delivery of siRNA. Acc. Chem. Res. 45, 961–970 (2012).
Sizovs, A. et al. Poly(trehalose): sugar-coated nanocomplexes promote stabilization and effective polyplex-mediated siRNA delivery. J. Am. Chem. Soc. 135, 15417–15424 (2013).
Gandra, N., Wang, D. D., Zhu, Y. & Mao, C. Virus-mimetic cytoplasm-cleavable magnetic/silica nanoclusters for enhanced gene delivery to mesenchymal stem cells. Angew. Chem. Int. Ed. 52, 11278–11281 (2013).
Singh, R. et al. Binding and condensation of plasmid DNA onto functionalized carbon nanotubes: toward the construction of nanotube-based gene delivery vectors. J. Am. Chem. Soc. 127, 4388–4396 (2005).
Singh, N. et al. Effect of nanoparticle conjugation on gene silencing by RNA interference. J. Am. Chem. Soc. 132, 8241–8243 (2010).
Mintzer, M. A. & Simanek, E. E. Nonviral vectors for gene delivery. Chem. Rev. 109, 259–302 (2009).
Savic, R., Luo, L., Eisenberg, A. & Maysinger, D. Micellar nanocontainers distribute to defined cytoplasmic organelles. Science 300, 615–618 (2003).
Roy, I. et al. Optical tracking of organically modified silica nanoparticles as DNA carriers: a nonviral, nanomedicine approach for gene delivery. Proc. Natl. Acad. Sci. USA 102, 279–284 (2005).
Jung, J. et al. Selective inhibition of human brain tumor cells through multifunctional quantum-dot-based siRNA delivery. Angew. Chem. Int. Ed. 49, 103–107 (2010).
Liu, Y. et al. ZnO quantum dots-embedded collagen/polyanion composite hydrogels with integrated functions of degradation tracking/inhibition and gene delivery. J. Mater. Chem. 22, 512–519 (2012).
Kim, J. et al. Transfection and intracellular trafficking properties of carbon dot-gold nanoparticle molecular assembly conjugated with PEI-pDNA. Biomaterials 34, 7168–7180 (2013).
Chen, A. A., Derfus, A. M., Khetani, S. R. & Bhatia, S. N. Quantum dots to monitor RNAi delivery and improve gene silencing. Nucleic Acids Res. 33, e190 (2005).
Liu, L. et al. Nanoparticles from block copolymer encapsulating Re(phen) complexes as bifunctional agents for cell imaging and gene transfection. Chem. Commun. 6759–6761 (2009).
Yu, B. et al. A luminescent tetranuclear ruthenium (II) complex as a tracking non-viral gene vector. Chem. Commun. 49, 810–812 (2013).
Liu, Q. et al. Sub-10 nm hexagonal lanthanide-doped NaLuF4 upconversion nanocrystals for sensitive bioimaging in vivo. J. Am. Chem. Soc. 133, 17122–17125 (2011).
Ju, Q. et al. Amine-functionalized lanthanide-doped KGdF4 nanocrystals as potential optical/magnetic multimodal bioprobes. J. Am. Chem. Soc. 134, 1323–1330 (2012).
Park, J. Y. et al. Paramagnetic ultrasmall gadolinium oxide nanoparticles as advanced T1 MRI contrast agent: account for large longitudinal relaxivity, optimal particle diameter and in vivo T1 MR images. ACS NANO 3, 3663–3669 (2009).
Liu, J. et al. Passive tumor targeting of renal-clearable luminescent gold nanoparticles: long tumor retention and fast normal tissue clearance. J. Am. Chem. Soc. 135, 4978–4981 (2013).
Smith, A. M., Duan, H., Mohs, A. M. & Nie, S. Bioconjugated quantum dots for in vivo molecular and celluar imaging. Adv. Drug Deliv. Rev. 60, 1226–1240 (2008).
Marks, K. M. & Nolan, G. P. Chemical labeling strategies for cell biology. Nat. Methods 3, 591–596 (2006).
Feng, G. et al. Ultrabright organic dots with aggregation-induced emission characteristics for cell tracking. Biomaterials 35, 8669–8677 (2014).
Resch-Genger, U., Grabolle, M., Cavaliere-Jaricot, S., Nitschke, R. & Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 5, 763–775 (2008).
Liu, L. et al. Dinuclear metal(II) complexes of polybenzimidazole ligands as carriers for DNA delivery. Biomaterials 31, 1380–1391 (2010).
Yin, J. et al. The effect of a nuclear locatization sequence on transfection efficacy of genes delivered by cobalt(II)-polybenzimidazole complexes. Biomaterials 33, 7884–7894 (2012).
Dong, X. et al. Reversible DNA condensation induced by a tetranuclear nickel(II) complex. Chem. Eur. J. 16, 14181–14189 (2010).
Kikuchi, T., Sato, S., Fujita, D. & Fujita, M. Stepwise DNA condensation by a histone-mimic peptide-coated M12L24 spherical complex. Chem. Sci. 5, 3257–3260 (2014).
Malina, J., Farrell, N. P. & Brabec, V. DNA condensing effects and sequence selectivity of DNA binding of antitumor noncovalent polynuclear platinum complexes. Inorg. Chem. 53, 1662–1671 (2014).
Newkome, G. R., He, E. & Moorefield, C. N. Suprasupermolecules with novel properties: metallodendrimers. Chem. Rev. 99, 1689–1746 (1999).
Hwang, S.-H., Shreiner, C. D., Moorefield, C. N. & Newkome, G. R. Recent progress and applications for metallodendrimers. New J. Chem. 31, 1192–1217 (2007).
Gingras, M., Raimondo, J. M. & Chabre, Y. M. Cleavable dendrimers. Angew. Chem. Int. Ed. 46, 1010–1017 (2007).
Govender, P., Therrien, B. & Smith, G. S. Bio-metallodendrimers – emerging strategies in metal-based drug design. Eur. J. Inorg. Chem. 19, 2853–2862 (2012).
Astruc, D., Boisselier, E. & Ornelas, C. Dendrimers designed for functions: from physical, photophysical and supramolecular properties to applications in sensing, catalysis, molecular electronics, photonics and nanomedicine. Chem. Rev. 110, 1857–1959 (2010).
Astruc, D. Electron-transfer processes in dendrimers and their implication in biology, catalysis, sensing and nanotechnology. Nat. Chem. 4, 255–267 (2012).
Wang, Y., Rapakousiou, A., Ruiz, J. & Astruc, D. Metalation of polyamine dendrimers with ethynylcobalticenium for the construction of mono- and heterobimetallic polycationic metallodendrimers. Chem. Eur. J. 20, 11176–11186 (2014).
Fréchet, J. M. Dendrimers and supramolecular chemistry. Proc. Natl. Acad. Sci. USA 99, 4787–4787 (2002).
Xu, W., Zuo, J., Wang, L., Ji, L. & Chao, H. Dinuclear ruthenium(II) polypyridyl complexes as single and two-photon luminescence cellular imaging probes. Chem. Commun. 50, 2123–2125 (2014).
Zhang, P. et al. A dinuclear ruthenium(II) complex as a one- and two- photon luminescent probe for biological Cu2+ detection. Chem. Eur. J. 19, 15494–15503 (2013).
Zhang, P. et al. RuNH2@AuNPs as two-photon luminescent probes for thiols in living cells and tissues. Biomaterials 35, 9003–9011 (2014).
Baggaley, E. et al. Dinuclear ruthenium(II) complexes as two-photon, time-resolved emission microscopy probes for cellular DNA. Angew. Chem. Int. Ed. 53, 3367–3371 (2014).
W. R. Zipfel, R. M. Willams, W. W. Webb, Nonlinear magic: multiphoton microscopy in the biosciences. Nat. Biotechnol. 21, 1369–1377 (2003).
U. K. Tirlapur, K. König, Targeted transfection by femtosecond laser. Nature 418, 290–291 (2002).
Yamada, M., Tanaka, Y., Yoshimoto, Y., Kuroda, S. & Shimao, I. Synthesis and properties of diamino-substituted dipyrido [3,2-a:2’,3’-c] phenazine. Bull. Chem. Soc. Jpn. 65, 1006–1011 (1992).
Sullivan, B. P., Salmon, D. J. & Meyer, T. J. Mixed phosphine 2,2’-bipyridine complexes of ruthenium. Inorg. Chem. 17, 3334–3341 (1978).
Juris, A. et al. Ruthenium(II) polypyridine complexes: photophysics, photochemistry, electrochemistry and chemiluminescence. Coord. Chem. Rev. 84, 85–277 (1988).
Xu, C. & Webb, W. Two-photon fluorescence excitation cross sections of biomolecular probes from 690 to 960 nm. J. Opt. Soc. Am. B 13, 481–491 (1996).
Ji, C., Zhang, L. Y., Hou, X. M., Dou, S. X. & Wang, P. Y. Effect of cisplatin on the flexibility of linear DNA. Chinese Phys. Lett. 28, 068702 (2011).
Acknowledgements
This work was supported by the 973 program (Nos. 2014CB845604 and 2015CB856301), the National Science Foundation of China (Nos. 21172273, 21171177, 21471164 and J1103305) and the Program for Changjiang Scholars and Innovative Research Team in University of China (No. IRT1298).
Author information
Authors and Affiliations
Contributions
H.C. conceived the idea and directed the work. K.Q. performed the experimentswith help from B.Y., H. H., P. Z., L. J. and Y.C. S.Z. prepared the 2kb DNA. J.H. drew the schematic representation. All authors contributed to data analysis and manuscript writing.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Qiu, K., Yu, B., Huang, H. et al. A dendritic nano-sized hexanuclear ruthenium(II) complex as a one- and two-photon luminescent tracking non-viral gene vector. Sci Rep 5, 10707 (2015). https://doi.org/10.1038/srep10707
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep10707
This article is cited by
-
Ruthenium-containing supramolecular nanoparticles based on bipyridine-modified cyclodextrin and adamantyl PEI with DNA condensation properties
Nanoscale Research Letters (2018)
-
Self-assembly of c-myc DNA promoted by a single enantiomer ruthenium complex as a potential nuclear targeting gene carrier
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
