
Abstract
A detailed study of the valence state and distribution of the AuCl3/AC catalyst during the acetylene hydrochlorination deactivation process is described and discussed. Temperature-programmed reduction and X-ray photoelectron spectral analysis indicate that the active Au3+ reduction to metallic Au0 is one reason for the deactivation of AuCl3/AC catalyst. Transmission electron microscopy characterization demonstrated that the particle size of Au nano-particles increases with increasing reaction time. The results indicated that metallic Au0 exhibits considerable catalytic activity and that Au nano-particle aggregation may be another reason for the AuCl3/AC catalytic activity in acetylene hydrochlorination.
Similar content being viewed by others

Introduction
Acetylene hydrochlorination is an important process in the industrial production of vinyl chloride monomer, which is the primary raw material used to synthesize polyvinyl chloride, especially in China, because of the vast domestic coal resources and the increasing cost of petroleum1. HgCl2 supported on activated carbon is a common catalyst for industrial acetylene hydrochlorination. However, the HgCl2 catalyst is toxic and harmful to human health. Therefore, the exploitation of non-mercury catalysts to replace HgCl2 is of significant importance for the survival and development of the PVC industry through the acetylene route.
Numerous metal complexes, including Bi3+, Pt6+, Pt4+ and Pd2+, have recently been applied as candidate catalysts for acetylene hydrochlorination2,3,4,5. Hutchings6,7 reported that Au-based catalysts are the optimal metal chloride catalysts for acetylene hydrochlorination. However, the Au-based catalyst is easily deactivated over the course of the reaction.
Many investigations have attempted to elucidate the deactivation mechanism of AuCl3/AC catalyst in acetylene hydrochlorination. Hutchings proposed that the reduction of Au3+ to Au0 is the primary deactivation pathway under the operating condition of acetylene hydrochlorination8,9. Hutchings further disclosed the reason for active Au3+ reduction in acetylene hydrochlorination and proposed that acetylene was more easily adsorbed by AuCl3 catalyst compared with the hydrogen chloride species and that the strong adsorption of acetylene caused the reduction of Au3+ 10. Shen et al. reported similar results. These researchers determined that the initial coordination of hydrogen chloride with AuCl3 produced a calculated energy of -23.90 kJ mol−1, whereas acetylene in the vacant coordination site of AuCl3 generates a relative energy of -66.65 kJ mol−1. Therefore, the adsorption of acetylene on AuCl3 is relatively stronger than that of hydrogen chloride11. Zhang et al. suggested a detailed deactivation mechanism of AuCl3 catalyst for acetylene hydrochlorination. These researchers determined that the electron in the p orbital of acetylene transfers to the unoccupied molecular orbital of Au3+, thereby causing the reduction of Au3+ to its low-valence state via the loss of its Cl atoms12.
From the above-described literature, the reduction of active Au3+ ions to metallic Au0 is the primary reason for the AuCl3/AC catalyst deactivation in acetylene hydrochlorination. However, all of these investigators ignored an obvious fact. Au nano-particles are present in the deactivated catalyst. Thus, Au nano-particle aggregation may play a role in the AuCl3 catalyst deactivation because the particle size of Au nano-particles significantly affects catalytic performance, as described in many reports on heterogeneous catalysis13,14,15. In our previous work, we observed that the AuCl3 particle size after acetylene hydrochlorination is notably large16 and metallic Au0 (3-10) clusters may exhibit considerable catalytic activity based on the results of simulations using density functional theory17. Therefore, Au nano-particle agglomeration may also influence the catalytic performance of AuCl3 catalyst in acetylene hydrochlorination.
In this paper, we investigate the relationship between the metallic Au0 particle size and its catalytic performance to elucidate the deactivation mechanism of AuCl3/AC for acetylene hydrochlorination. A series of characterizations was performed to determine the valence state and distribution of the AuCl3/AC catalysts in various stages of catalyst life.
Experimental Section
Materials
Activated carbon (marked as AC, neutral, 40-60 mesh), HAuCl4·4H2O (with 47.8% Au content), C2H2 (gas, 98%) and HCl (gas, 99%) were used in this study.
Catalyst preparation
The AuCl3/AC catalysts were prepared using an incipient wetness impregnation technique that used aqua regia as a solvent, as described in the literature18. The gold precursor, HAuCl·4H2O, was dissolved in aqua regia [3:1 HCl (Fisher, 32%): HNO3 (Fisher, 70%) by volume, 6.4 mL] and the solution was added dropwise with stirring to the activated carbon support (3.00 g) to obtain a catalyst with a final metal loading of 1 wt.%. The mixture was maintained for 24 h at room temperature and then the product was dried for 18 h at 140 °C before being used as a catalyst.
The Au/AC catalyst (1 wt.% Au) was prepared using a colloidal deposition technique similar to that in the literature19, with a few modifications. First, 100 mL of distilled water was mixed with 3.2 mL of 2.43 × 10−2 M AuCl4- solution and then 5 mL of 0.1 M fresh NaBH4 solution was added dropwise. The solution was stirred for 2 h. Next, activated carbon (1.50 g) was added and then the pH value was adjusted to 1.0 using 1.0 M HCl. Subsequently, the solution was vigorously stirred overnight. Finally, the mixture was washed with distilled water and then dried in a vacuum oven at 80 °C to yield the Au/AC catalyst. Two samples were calcined for 4 h at 300 °C and at 800 °C (5 °C /min), the obtained samples were labeled as Au/AC-300 and Au/AC-800, respectively.
Catalyst characterization
X-ray diffractometer data were collected using a Bruker D8 advanced X-ray diffractometer equipped with a Cu-Kα irradiation source (λ = 1.5406 Å) operating at 40 kV and 40 mA, with data collected over the 2θ scanning range between 20° and 90°. The morphologies of the samples were examined via transmission electron microscopy using a JEM 2010 electron microscope operating at an accelerating voltage of 200 kV, with a line resolution of 0.14 nm and a point-to-point resolution of 0.23 nm. Temperature-programmed reduction was performed using a similar Micromeritic ASAP 2720 apparatus equipped with a TCD detector. The reducing gas was 10% H2 in Ar, with a flow rate of 40 mL min−1. The temperature range was from 50 °C to 400 °C, with a heating rate of 10 °C min−1. X-ray photoelectron spectrum data were recorded using an Axis Ultra spectrometer with a monochromatized Al-Kα X-ray source (225 W), a minimum energy resolution of 0.48 eV (Ag 3d5/2) and a minimum X-ray photoelectron spectrum analysis area of 15 μm.
Catalytic performance evaluation
The catalytic performance during acetylene hydrochlorination was evaluated in a fixed-bed glass microreactor (i.d. of 10 mm) operating just above atmospheric pressure. A CKW 1100 temperature controller (Chaoyang Automation Instrument Factory, Beijing, China) regulated the reactor temperature. The reactor was purged with nitrogen to remove water and air in the reaction system before the reaction process. Hydrogen chloride gas was passed through the reactor at a flow rate of 20 mL min−1 to activate the catalyst. After the reactor was heated to 180 °C, acetylene (24.3 mL min−1) and hydrogen chloride (29.4 mL min−1) were fed through the heated reactor containing 2 mL of catalyst, with a gas hourly space velocity of 870 h−1. The reaction products were analyzed using a gas chromatograph (GC-2014C) produced by Shimadzu International Trading Company Ltd. (Shanghai). Analysis conditions: chromatographic column type of 2 m × Φ 4 mm, stuffing of GDX-301, column temperature of 120 °C, FID detector, detector and vaporizer temperature of 150 °C and injection volume of 60 μL. The conversion of acetylene (XA) and the selectivity to vinyl chloride monomer (SVC) as the criteria of catalytic performance20 were defined as the equations (1) and (2), respectively.


In the equations,  is defined as the volume fraction of acetylene in the raw gas,
is defined as the volume fraction of acetylene in the raw gas,  is defined as the volume fraction of remaining acetylene in the product gas and
is defined as the volume fraction of remaining acetylene in the product gas and  is the volume fraction of vinyl chloride in the product gas.
is the volume fraction of vinyl chloride in the product gas.
Results and Discussion
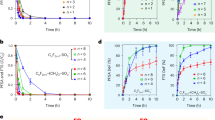
To accelerate the deactivation process of Au-based catalysts, we applied both a gas hourly space velocity and a reaction temperature that were higher than those reported in the literature21. The catalysts were tested under fixed reaction conditions (C2H2/HCl = 1.15:1, gas hourly space velocity = 870 h−1, reaction temperature = 180 °C). The result is illustrated in Fig. 1A. The catalyst initially exhibited low conversion (ca. 16.71%), followed by an increase in activity after approximately 2 h and then reached ca. 84.61% conversion. From the initial stage of the reaction (~ 2 h), there exists an activation period for the fresh AuCl3/AC catalyst during the acetylene hydrochlorination reaction. This result indicated that the most active sites for acetylene hydrochlorination are formed during this period. We characterized the AuCl3/AC catalyst during the initial stages of the reaction (~ 0.5 h, 1 h and 1.5 h) by X-ray photoelectron spectrum experiments and the results are listed in Fig. 2 and Table 1. From the results of Table 1, the compositions of Au3+ species in AuCl3/AC-0.5, AuCl3/AC-1 and AuCl3/AC-1.5 are observed to be 42.39%, 38.58% and 36.82%, respectively. Note that the content of the Au3+ species decreased, while the catalytic activity increased. This result indicated that Au3+ was not the only active site for acetylene hydrochlorination. The acetylene conversion decreased to ca. 68.70% after reacting for 4 h and then the rate of acetylene conversion of the AuCl3/AC catalyst decreased to ca. 9.96% after 8 h and the selectivity to vinyl chloride monomer of all AuCl3/AC catalysts was maintained at 99.99%, as shown in Fig. 1B. The results indicated that the AuCl3/AC catalyst was easily deactivated with increasing reaction time on the stream under these reaction conditions. Thus, we investigated the valence state and distribution of the catalyst at various stages of catalyst life. The catalyst was denoted as AuCl3/AC-x (where x represents the reaction time of 0, 2, 4, 6 and 8 h). The actual Au amount in AuCl3/AC-0, AuCl3/AC-2, AuCl3/AC-4, AuCl3/AC-6 and AuCl3/AC-8 catalysts was 1.09 wt.%, 0.98 wt.%, 1.02 wt.%, 1.04 wt.% and 1.01 wt.%, respectively, as determined by inductively coupled plasma-atomic emission spectrometry.

Acetylene conversion (A) and vinyl chloride monomer selectivity (B) during acetylene hydrochlorination catalyzed by (a) AuCl3/AC-0, (b) AuCl3/AC-2, (c) AuCl3/AC-4, (d) AuCl3/AC-6 and (e) AuCl3/AC-8. Reaction conditions: temperature = 180 °C, C2H2 gas hourly space velocity = 870 h−1, feed volume ratio VHCl/VC2H2 = 1/1.15.

High-resolution X-ray photoelectron spectrum for Au 4f of (a) AuCl3/AC-0.5, (b) AuCl3/AC-1 and (c) AuCl3/AC-1.5.
X-ray diffractometer patterns of the representative AuCl3/AC-x catalysts are shown in Fig. 3. Two diffraction peaks in the pattern of AuCl3/AC-0 catalyst appear at 2θ = 24.4° and 43.7°, corresponding to the planes of (0 0 2) and (1 0 1) , respectively, of the AC support22. No obvious diffraction of Au3+ or metallic Au0 is observed in the fresh AuCl3/AC-0 catalyst, indicating that active Au3+ components are highly dispersed on the surface of the AC support. For AuCl3/AC-2, AuCl3/AC-4, AuCl3/AC-6 and AuCl3/AC-8 catalysts, obvious diffraction peaks appear at 38.5°, 44.7°, 64.8° and 77.9° (2θ), respectively, corresponding to the planes (1 1 1), (2 0 0), (2 2 0) and (3 1 1), i.e., the diffraction peaks of metallic Au0 (JCPDS, No. 4-0784), for 38.5°, 44.7°, 64.8° and 77.9° (2θ), respectively23. The X-ray diffractometer results show that Au3+ is reduced to metallic Au0 in acetylene hydrochlorination. Therefore, the reduction of an active Au3+ component is one reason for the deactivation of the AuCl3/AC catalyst in the acetylene hydrochlorination, which is consistent with the literature results8,10.

X-ray diffractometer patterns of (a) AuCl3/AC-0, (b) AuCl3/AC-2, (c) AuCl3/AC-4, (d) AuCl3/AC-6 and (e) AuCl3/AC-8.
In view of the above results, there still must be detailed investigations on the deactivation mechanism to make it clear whether a change of surface gold oxidation state was responsible for the deactivation for acetylene hydrochlorination. To correlate the bulk changes of the surface Au3+ in catalysts with the surface of the gold oxidation state, X-ray photoelectron spectroscopy was systematically performed for all AuCl3/AC-x catalysts. The deconvolution results in Fig. 4 indicate that each gold species shows two peaks, the gold species with the binding energy at 84.1 eV and 87.7 eV, due to Au 4f7/2 and the Au 4f5/2 spectrum of metallic Au0 24; the spectral peaks at binding energies of 85.2 eV and 88.6 eV are assigned with Au3+. As shown in Fig. 4, the gold species on the surface of the catalysts exists as metallic Au and Au3+ states. The relative content of Au species in the fresh air and in the used catalysts, based on the deconvolution of X-ray photoelectron spectroscopy are tabulated in Table 2. As listed in Table 2, the amounts of gold species in the used catalysts differ greatly from those in the fresh one. Compared with the AuCl3/AC-0 catalyst, the amounts of Au0 increase from 50.55% to 65.47%, while the contents of Au3+ decrease after undergoing the reaction for 2 h. With increasing reaction time on the stream, the surface composition of approximately 89.38% of the Au0 species and 10.62% of the Au3+ species after 8 h, indicating that a great amount of Au3+ can be reduced to Au0 in the acetylene atmosphere. Therefore, the gold species on the catalysts surface exists as metallic Au0 and Au3+ states in acetylene hydrochlorination, while, approximately 54.65% of the Au3+ component is reduced to Au0 after reaction, which is one reason for the deactivation of AuCl3/AC catalyst in acetylene hydrochlorination.

High-resolution X-ray photoelectron spectrum for Au 4f of (a) AuCl3/AC-0, (b) AuCl3/AC-2, (c) AuCl3/AC-4, (d) AuCl3/AC-6 and (e) AuCl3/AC-8.
To further quantify of the bulk Au3+ amount and the gold oxidation state during acetylene hydrochlorination reaction, temperature-programmed reduction was performed to monitor the amount of active Au3+ by integrating the reduction peak area of these AuCl3/AC-x catalysts at different reaction times during acetylene hydrochlorination. As shown in Fig. 5, all AuCl3/AC-x catalysts exhibit one characteristic reduction band between 250 °C and 350 °C with the peak centered at 277 °C, which is attributed to Au3+ reduction25. Determined from the reduction peak of temperature-programmed reduction, the Au3+ content was found to follow the same general trend obtained from X-ray photoelectron spectroscopy. Hence, the relative amounts of Au3+ decrease with increasing reaction time on stream, which is consistent with the order of the catalytic performance of the catalysts (shown in Fig. 1A). Moreover, the Au3+ content in these catalysts is well correlated with the acetylene conversion approximated by a single line, as shown in Fig. 6.

Temperature-programmed reduction profiles of (a) AuCl3/AC-0, (b) AuCl3/AC-2, (c) AuCl3/AC-4, (d) AuCl3/AC-6 and (e) AuCl3/AC-8.

The above results indicate that approximately 13.59% of the Au3+ component is reduced to metallic Au0 when the reaction time is 2 h. If the Au3+ component is the only active site for acetylene hydrochlorination and the acetylene conversion of AuCl3/AC-0 should be higher than that of the AuCl3/AC-2 catalyst. However, we observed a contradictory phenomenon in Fig. 1. The acetylene conversion of AuCl3/AC-2 (84.61%) is significantly higher than that of AuCl3/AC-0 (16.71%), as shown in Fig. 1. Therefore, we hypothesize that Au3+ is not the only active site for acetylene hydrochlorination and metallic Au0 may also be active for this reaction.
To prove this hypothesis, we synthesized carbon support Au nano-particle with NaBH4 as a reducing agent and compared its catalytic performance for acetylene hydrochlorination with that of the AuCl3/AC catalyst. NaBH4 is a very strong reducing agent; thus, only metallic Au0 is present in the prepared Au/AC catalyst, as reported in the literature26. The catalytic performance of Au/AC catalyst for acetylene hydrochlorination was evaluated under a fixed reaction condition (C2H2/HCl = 1:1.15, gas hourly space velocity = 870 h−1, reaction temperature = 180 °C) compared with the AuCl3/AC catalyst. Notably, the acetylene conversion of AuCl3/AC is relatively higher than that in Fig. 1 because of the different reaction conditions. The acetylene conversion of the Au/AC catalyst is 57.1%, which is considerably lower than that of AuCl3/AC catalyst in Fig. 7. This result indicates that Au3+ is more active than metallic Au0, consistent with literature25,27. However, the catalytic activity of Au/AC is still higher than that of the AuCl3/AC-8 catalyst. If the reduction of Au3+ to Au0 is the only reason for the deactivation of AuCl3/AC catalyst, then the AuCl3/AC-8 catalytic activity should be close to that of Au/AC. Therefore, the possibility exists for another cause of the deactivation of AuCl3/AC catalyst for acetylene hydrochlorination, apart from the reduction of the active Au3+ component.

Acetylene conversion during acetylene hydrochlorination catalyzed by (a) AuCl3/AC and (b) Au/AC. Reaction conditions: temperature = 180 °C, C2H2 gas hourly space velocity = 870 h−1 and feed volume ratio VHCl/VC2H2 = 1.15.
Representative transmission electron microscopy images of the AuCl3/AC-x catalysts are presented in Fig. 8 to determine the distribution of Au nano-particles in the acetylene hydrochlorination process. Fig. 8(a) shows no obvious presence of Au0 particles in the AuCl3/AC-0 catalyst, which demonstrates that the Au element is found in the form of Au3+ ion, which is invisible in the transmission electron microscopy micrographs. The X-ray diffractometer results confirm the transmission electron microscopy measurements and indicate that the AuCl3/AC-0 catalyst contains Au3+ only. While the X-ray photoelectron spectroscopy experiment exhibits an obvious Au0 peak in the sample, because the quantitative assessment of the gold oxidation state by X-ray photoelectron spectroscopy characterization alone is limited, the final state effects associated with particle size could significantly disturb the X-ray photoelectron spectroscopy features of Au28. For the AuCl3/AC-2, AuCl3/AC-4 and AuCl3/AC-6 catalysts, the average particle sizes of the Au0 species are 22.1, 34.2, 66.1 and 81.2 nm, respectively. The particle size of Au nano-particle increases with increasing reaction time on stream, which indicates that the Au nano-particles form aggregates during acetylene hydrochlorination. As previously discussed, metallic Au0 exhibits considerable catalytic activity for acetylene hydrochlorination and the catalytic activity of the AuCl3/AC-x catalyst decreases with the reaction time increment during the acetylene hydrochlorination process. Therefore, Au nano-particle aggregation is another reason for the deactivation of AuCl3/AC catalyst in acetylene hydrochlorination.

Transmission electron microscopy images of the catalysts: (a) AuCl3/AC-0, (b) AuCl3/AC-2, (c) AuCl3/AC-4, (d) AuCl3/AC-6 and (e) AuCl3/AC-8.
We prepared Au/AC catalysts with different particle sizes by subsequent heat-treatment under a nitrogen atmosphere at 300 °C and 800 °C for 4 h to support this viewpoint further. The particle sizes of the Au/AC, Au/AC-300 and Au/AC-800 catalysts are 21.3, 40.6 and 83.6 nm, respectively, as shown in Fig. 9. The catalytic activity of the Au catalysts decreases in the order of Au/AC > Au/AC-300 > Au/AC-800 (as shown in Fig. 10). The result determined that the Au/AC catalyst with larger particle size exhibited lower catalytic activity for acetylene hydrochlorination. This result also provides evidence that Au nano-particle aggregation is one of the reasons for the deactivation of the AuCl3/AC catalyst in acetylene hydrochlorination.

Transmission electron microscopy images of (a) Au/AC, (b) Au/AC-300 and (c) Au/AC-800.

Acetylene conversion during acetylene hydrochlorination catalyzed by (a) Au/AC, (b) Au/AC-300 and (c) Au/AC-800. Reaction conditions: temperature = 180 °C, C2H2 gas hourly space velocity = 870 h−1 and feed volume ratio VHCl/VC2H2 = 1.15.
This paper reported the valence state and distribution of the AuCl3/AC catalyst at various stages of catalyst life for acetylene hydrochlorination. Metallic Au0 was found to exhibit considerable catalytic activity for acetylene hydrochlorination; as a result, Au nano-particle aggregation is another reason for the deactivation of AuCl3/AC catalyst, apart from the reduction of Au3+ component. Au nano-particle inhibition may be a development strategy for the exploration of a stable Au-based catalyst for acetylene hydrochlorination.
Additional Information
How to cite this article: Dai, B. et al. Effect of Au nano-particle aggregation on the deactivation of the AuCl3/AC catalyst for acetylene hydrochlorination. Sci. Rep. 5, 10553; doi: 10.1038/srep10553 (2015).
References
Zhang, J. L., Liu, N., Li. W. & Dai, B. Progress on cleaner production of vinyl chloride monomers over non-mercury catalysts. Front. Chem. Sci. Eng. 5, 514–520 (2011).
Smith, D. M., Walsh, P. M., & Slager, T. L. Studies of silica-supported metal chloride catalysts for the vapor-phase hydrochlorination of acetylene. J. Catal. 11, 113–130 (1968).
Mitchenkoa, S. A., Khomutov, E. V., Shubin, A. & Shul’ga, Y. M. Catalytic hydrochlorination of acetylene by gaseous HCl on the surface of mechanically pre-activated K2PtCl6 salt. J. Mol. Catal. A: Chem. 212, 345–350 (2004).
Mitchenkoa, S. A., Krasnyakova, T. V., Mitchenkoa, R. S. & Korduband, A. N. Acetylene catalytic hydrochlorination over powder catalyst prepared by pre-milling of K2PtCl4 salt. J. Mol. Catal. A: Chem. 275, 101–108 (2007).
Song, Q. L., Wang, S. J., Shen, B. X. & Zhao, J. G. Palladium-Based Catalysts for the Hydrochlorination of Acetylene: Reasons for Deactivation and Its Regeneration. Pet. Sci. Technol. 28, 1825–1833 (2010).
Hutchings, G. Vapor Phase Hydrochlorination of Acetylene: Correlation of Catalytic Activity of Supported Metal Chloride Catalysts. J. Catal. 96, 292–295 (1985).
Nkosi, B., Coville, N. & Hutchings, G. Vapour Phase Hydrochlorination of Acetylene with Group VIII and IB Metal Chloride Catalysts. J. Chem. Soc., Chem. Commun. 1, 71–72 (1988).
Nkosi, B. et al. Hydrochlorination of acetylene using gold catalysts: A study of catalyst deactivation. J. Catal. 128, 366–377 (1991).
Conte, M., Carley, A. & Hutchings, G. Reactivation of a Carbon-supported Gold Catalyst for the Hydrochlorination of Acetylene. Catal. Lett. 124, 165–167 (2008).
Conte, M. et al. Hydrochlorination of acetylene using a supported gold catalyst: A study of the reaction mechanism. J. Catal. 250, 231–239 (2007).
Wang, S. J., Shen, B. X. & Song, Q. L. Kinetics of Acetylene Hydrochlorination over Bimetallic Au–Cu/C Catalyst. Catal. Lett. 134, 102–109 (2010).
Zhang, J. L., He, Z. H., Li, W. & Han, Y. Deactivation mechanism of AuCl3 catalyst in acetylene hydrochlorination reaction: a DFT study. RSC Adv. 2, 4814–4821 (2012).
Lopez, N. et al. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 223, 232–235 (2004).
Menezes, W. G., Zielasek, V., Thiel, K., Hartwig, A. & Bäumer, M. Effects of particle size, composition and support on catalytic activity of AuAg nanoparticles prepared in reverse block copolymer micelles as nanoreactors. J. Catal. 299, 222–231 (2013).
Ghosh, P., Camellone, M. F. & Fabris, S. Fluxionality of Au Clusters at Ceria Surfaces during CO Oxidation: Relationships among Reactivity, Size, Cohesion and Surface Defects from DFT Simulations. J. Phys. Chem. Lett. 4, 2256–2263 (2013).
Li, X. Y., Dai, B. & Zhu, M. Y. AuCl3 on polypyrrole-modified carbon nanotubes as acetylene hydrochlorination catalysts. Appl. Catal. B: Environ. 142-143, 234–240 (2013).
Wang,Y., Zhu, M.Y., Kang, L.H & Dai, B. Neutral Aun (n = 3-10) clusters catalyze acetylene hydrochlorination: a density functional theory study. RSC Adv. 4, 38466–38473 (2014).
Conte, M. et al. Hydrochlorination of acetylene using supported bimetallic Au-based catalysts. J. Catal. 257, 190–198 (2008).
Porta, F., Prati, L., Rossi, M., Coluccia, S., & G. martra. Hydrochlorination of acetylene using supported bimetallic Au-based catalysts. Catal.Today 61, 165 (2000).
Wang, S. J., Shen, B. X., Zhao, J. G. & Liu, J. C. Deactivation of PdCl2/C Catalyst in Hydrochlorination of Acetylene. Petrochem. Technol. 38, 249–253 (2009).
Zhang, H.Y., Dai, B., Wang, X.G., Xu, L.L. & Zhu, M.Y. Hydrochlorination of acetylene to vinyl chloride monomer over bimetallic Au–La/SAC catalysts. J. Ind. Eng. Chem. 18, 49–54 (2012).
Qian, H. S. et al. Non-catalytic CVD preparation of carbon spheres with a specific size. Carbon 42, 761–766 (2004).
Chen, Y. W., Chen, H. J. & Lee, D. S. Au/Co3O4–TiO2 catalysts for preferential oxidation of CO in H2 stream. J. Mol. Catal. A: Chem. 363, 470–480 (2012).
Jaramillo, T. F., Baeck, S. H., Cuenya, B. R. & McFarland, E. W. Catalytic Activity of Supported Au Nanoparticles Deposited from Block Copolymer Micelles. J. Am. Chem. Soc. 125, 7148–7149 (2003).
Conte, M. et al. Modifications of the metal and support during the deactivation and regeneration of Au/C catalysts for the hydrochlorination of acetylene. Catal. Sci. Technol. 3, 128–134 (2013).
Tsunoyama, H., Sakurai, H., Negishi, Y. & Tsukuda, T. Size-Specific Catalytic Activity of Polymer-Stabilized Gold Nanoclusters for Aerobic Alcohol Oxidation in Water. J. Am. Chem. Soc. 127, 9374–9375 (2005).
Nkosi, B., Adams, M. D., Coville, N.J. & Hutchings, G. J. Hydrochlorination of acetylene using carbon-supported gold catalysts: A study of catalyst reactivation. J. Catal. 128, 378–386 (1991).
Huang, C. F., Zhu, M. Y., Kang, L. H., Li, X. Y. & Dai, B. Active carbon supported TiO2-AuCl3/AC catalyst with excellent stability for acetylene hydrochlorination reaction. Chem. Eng. J. 242, 69–75 (2014).
Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program, 2012CB720302) and the National Natural Science Funds of China (NSFC, 21366027, U1403294).
Author information
Authors and Affiliations
Contributions
B.D. and M.Y.Z. designed experiments. F.Y. and Q.Q.W. prepared samples and carried out characterization and catalyst activity test. M.Y.Z. and Q.Q.W. contributed to the analysis and discussion for the results. M.Y.Z. and Q.Q.W. wrote the paper. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dai, B., Wang, Q., Yu, F. et al. Effect of Au nano-particle aggregation on the deactivation of the AuCl3/AC catalyst for acetylene hydrochlorination. Sci Rep 5, 10553 (2015). https://doi.org/10.1038/srep10553
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep10553
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
