
Abstract
Rab27A regulates transport of lysosome-related organelles (LROs) and release of secretory granules in various types of cells. Here, we identified up-regulation of Rab27A during differentiation of osteoclasts (OCLs) from bone-marrow macrophages (BMMs), by DNA microarray analysis. Rab27A deficiency in OCLs, using small interfering RNA (siRNA) knockdown in RAW-D cell line or BMMs derived from ashen mice, which display genetic defects in Rab27A expression, induced multinucleated and giant cells. Upon stimulation with macrophage-colony stimulating factor (M-CSF) and receptor activator of nuclear factor kappa-B ligand (RANKL), essential cytokines for OCL differentiation, phosphorylation levels of extracellular signal-regulated kinase (Erk), proto-oncogene tyrosine-protein kinase (Src) and p-38 were slightly enhanced in ashen BMMs than in wild-type BMMs. The cell surface level of c-fms, an M-CSF receptor, was slightly higher in ashen BMMs than in wild-type BMMs and down-regulation of RANK, a RANKL receptor, was delayed. In addition to receptors, OCLs derived from ashen mice exhibited aberrant actin ring formation, abnormal subcellular localization of lysosome-associated membrane protein (LAMP2) and cathepsin K (CTSK) and marked reduction in resorbing activity. Thus, these findings suggest that Rab27A regulates normal transport of cell surface receptors modulating multinucleation and LROs in OCLs.
Similar content being viewed by others


Introduction
The Rab family of small GTPases mediates membrane trafficking events such as vesicle formation, vesicle movement and membrane fusion1. A member of this family, Rab27A, has been extensively studied2. Griscelli syndrome type 2 is a genetic disorder affecting Rab27A in humans and it is characterized by hypopigmentation of the skin and eyes, immunodeficiency3,4. Rab27A is widely expressed on secretory granules in various secretory cells, such as endocrine and exocrine cells and various leukocytes5. Rab27A is particularly involved in regulation of transport of “lysosome-related organelles (LROs)”6,7. LROs resemble morphologically lysosomes with electron-dense protein deposits and contain most lysosomal proteins and have a low luminal pH. However, they display many distinct morphological, functional and compositional characteristics8. LROs include the melanosomes in melanocytes9, lytic granules in lymphocytes3,10, delta granules in platelets11 and “secretory” lysosomes in osteoclasts (OCLs)12. Compared to the other LROs, the transport mechanisms of LROs in OCLs are not well known.
OCLs are multinucleated giant cells that mainly participate in bone resorption13,14. OCLs are formed by the fusion of mononuclear progenitors of the monocyte/macrophage lineage15. When OCLs undergo bone resorption, they form a specialized LRO, termed as the “ruffled border”16,17. The ruffled border generates an acidic extracellular microenvironment called as resorption lacuna, which is surrounded by an actin ring18. Several acid hydrolases are secreted in the resorption lacuna, which is formed on the bone surface19. Among these acid hydrolases, cathepsin K (CTSK) and tartrate-resistant acid phosphatase (TRAP, also known as ACP5) are highly expressed and secreted by differentiated OCLs. CTSK is a lysosomal cysteine protease that degrades type I collagen, the major component of bone matrix20,21,22. TRAP is a metallo-phosphoesterase involved in bone matrix degradation and removal of mannose 6-phosphate (Man-6-P) residues from acid hydrolases23,24. However, the detailed transport mechanisms of LROs in OCLs are yet to be elucidated. In addition, the extent of involvement of Rab27A in the transport of LROs in OCLs is still unclear.
In this study, we identified the up-regulation of Rab27A during OCL differentiation from bone-marrow macrophages (BMMs), using DNA microarray analysis. Moreover, we have demonstrated, by knock-down using small interfering RNA (siRNA) and the Rab27A-deficient ashen mice, that Rab27A regulates the transport of LROs and cell surface receptors, thereby modulating the cell size in OCLs.
Results
Expression of Rab27A increases during OCL differentiation
To identify a gene which regulates membrane trafficking during OCL differentiation, we performed DNA microarray analysis. Total RNA was obtained from BMMs treated with M-CSF (30 ng/ml) and RANKL (50 ng/ml) and cultured for 72 h on a plastic surface or a dentin slice. We observed that the OCLs cultured on the plastic surface were differentiated rapidly into OCL rather than on the dentin slice. Therefore, we compared the mRNA levels of OCLs cultured under the two different conditions. Of the total of 40,130 genes identified during the analysis, 1,363 were up-regulated and 881 genes were down-regulated. Indeed, OCL marker genes such as calcitonin receptor (CTR), cathepsin K (CTSK), transmembrane 7 superfamily member 4 (DC-STAMP), carbonic anhydrase 2 and TRAP were up-regulated (Supplementary Figure S1A). Among the several up-regulated genes, we focused on Rab27A, since Rab27A expression showed an increased, but that of Rab27B decreased during OCL differentiation.
We further measured the mRNA levels of Rab27A and Rab27B in MC3T3-E1 cells (murine osteoblastic precursor cell line), RAW-D cells (sub-clone of the murine macrophage cell line RAW264.7, which has a high capacity for differentiation into OCLs)25 and RANKL-stimulated RAW-D cells (OCLs). Quantitative real-time PCR analysis showed that the mRNA level of Rab27A in RAW-D cells was a 4.5-fold higher than that in MC3TC-E1 cells (Supplementary Figure S1B). Moreover, upon stimulation with RANKL, the Rab27A mRNA expression in mature OCLs was significantly increased compared to that in unstimulated RAW-D cells (Supplementary Figure S1B). However, the mRNA levels of Rab27B in RAW-D and OCLs were not detectable (Supplementary Figure S1B). Thus, we concluded that Rab27A expression was significantly increased during differentiation from macrophages into OCLs.
Rab27A is localized in lysosomes in RAW-D cells
We next determined the subcellular localization of Rab27A in RAW-D cells. Since the staining pattern of endogenous Rab27A in the OCL precursor cell RAW-D and RANKL-stimulated OCLs differentiated from RAW-D cells, with several anti-Rab27A antibodies was obscure, we analyzed overexpression of Rab27A-Green Fluorescent Protein (GFP) in RAW-D cells. We compared the localization of Rab27A against several organelle marker proteins. As shown in Supplementary Figure S2, the Rab27A-GFP protein was extensively colocalized with LAMP2 (late endosomes/lysosomes), but not with GM130 (Golgi) and calnexin (endoplasmic reticulum; ER). These results indicate that Rab27A is localized in the lysosomes and late endosomes in RAW-D cells.
Knockdown of Rab27A causes formation of multinucleated, giant OCLs
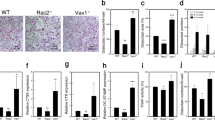
To explore the role of Rab27A during OCL differentiation, we performed knockdown by siRNA transfection in RANKL-stimulated RAW-D cells. We determined the knockdown efficiency of Rab27A in the RAW-D derived OCLs (Fig. 1A). Rab27A siRNA potently inhibited its expression level down to approximately 40% as compared to that obtained from a control siRNA (Fig. 1a). Under these conditions, Rab27A depletion induced considerably distinct morphological characteristics in OCLs such as multinucleated and giant cells, when both the cells were cultured for 72 h after stimulation with RANKL (Fig. 1b). A total nucleus number count in TRAP-positive multinucleated cells transfected with the control siRNA and Rab27A siRNA showed a higher nucleus number in Rab27A depleted OCLs, compared to that seen in the control OCLs (Fig. 1c). This suggests that the control cells largely remained as TRAP-negative mononuclear cells (Fig. 1c). However, the number of TRAP positive multinucleated OCLs was not different between the control and Rab27A knockdown OCLs (Fig. 1d). These results indicate that Rab27A depletion in OCLs induce multinucleation and larger cell formation.

Knockdown of Rab27A in RAW-D derived OCLs after stimulation with RANKL.
(a) Knockdown efficiency was measured by mRNA levels of Rab27A. After incubation with 10 p mol siRNA for 24 h, cells were cultured for additional for 5 days in the presence of RANKL (50ng/mL). (b) RAW-D cells were transfected with either a control siRNA or a siRNA specific for Rab27A. After stimulation with RANKL (50 ng/mL) for 72 h, OCLs were analyzed by TRAP-staining. (c) Total nucleus number of TRAP positive multinucleated OCLs, but not TRAP negative mononucleated cells following a 72 h culture, was counted per viewing field. ** P < 0.01. (D) The number of TRAP-positive multinucleated OCLs per viewing field was counted. Bar: 50 μm.
OCLs derived from Rab27A-deficient (ashen) mice displays multinucleation and giant cell formation
Given the importance of Rab27A in osteoclastogenesis, we compared the phenotypes of OCL differentiation in BMMs derived from ashen mice, which show genetic defects in Rab27A expression and control mice10,26. The BMMs were cultured with media containing M-CSF and RANKL, essential cytokines for OCL maturation, for 5, 6, 7 and 8 days. TRAP staining revealed that OCLs from ashen mice exhibited a markedly bigger size compared to OCLs from the wild-type mice (Fig. 2a). The number of TRAP-positive cells was higher in ashen mice than that of the wild-type mice at all the indicated days (Fig. 2b). Further, we counted the nuclear number of the OCLs following nuclear staining with DAPI (Fig. 2c). The OCLs from ashen mice containing more than 11 nuclei accounted for approximately 50% of the total number, whereas those from the wild-type did for only approximately 10% (Fig. 2d). A calculation of the cell area of OCLs from wild-type and ashen mice at 8 days showed a higher cell area in ashen mice than in wild-type mice (Fig. 2e).

Phenotypes of OCLs derived from control mice (wild-type) and ashen mice.
(a) BMMs derived from wild-type and ashen mice were cultured with M-CSF (10 ng/ml) and RANKL (50 ng/ml) for the indicated days. The cells were fixed and stained for TRAP. Bar: 50 μm. (b) The number of TRAP-positive multinucleated cells was counted at each indicated day. (c) The cells were fixed and stained with nuclear staining DAPI. Bar: 50 μm. (d) the nuclear numbers were counted and classified as 5 ~ 10 (white bar), 11 ~ 50 (gray bar), or more than 50 (black bar). (e) The individual cell area was analyzed by using the BZ analyzer software, dynamic cell count system (Keyence).
Partially enhanced maturation of ashen OCL
To examine the differences between wild-type and ashen OCLs, we compared mRNA levels of various OCL marker genes in wild-type and ashen OCLs. As shown in Fig. 3, quantitative real-time PCR revealed that mRNA levels of RANK, c-fms, CTR, DC-STAMP, OC-STAMP and CTSK were significantly higher in ashen OCLs compared to wild-type OCLs. However, the levels of integrin β3 and Src were indistinguishable in wild-type and ashen OCLs. (Fig. 3). These results indicate the partially enhancement of marker gene expression in ashen OCLs compared to wild-type OCLs.

Comparison of mRNA levels of various marker proteins OCLs from wild-type and ashen mice.
BMMs derived from wild-type and ashen mice were cultured with M-CSF (10 ng/ml) and RANKL (50 ng/ml) for 5 days. After isolation of mRNA in these cells, real-time PCR was performed. **P < 0.01, *P < 0.05 for the indicated comparisons.
Enhanced signaling of M-CSF and RANKL in OCLs from ashen mice
To investigate molecular mechanisms responsible for larger size of OCLs from ashen mice, we tested the signaling levels between wild-type and ashen BMMs, which are OCL precursor cells, by stimulation with M-CSF or RANKL. When stimulated with a concentration of M-CSF (10 ng/ml), the phosphorylation levels of Erk, p-38 and Src were slightly enhanced in ashen BMMs compared to wild-type cells (Fig. 4a). However, the phosphorylation levels of Akt were indistinguishable between wild-type and ashen BMMs (Fig. 4a). Upon stimulation with RANKL (50 ng/ml), the phosphorylation levels of Erk, p-38 and Akt in ashen BMMs were increased compared to wild-type cells (Fig. 4b). These results indicate that the enhanced signaling as a result of M-CSF and RANKL stimulation is probably one of the major factors responsible for larger size in ashen OCLs than in the wild-type OCLs.

Comparison of signaling levels of OCLs from wild-type and ashen mice.
(a) BMMs from wild-type and ashen mice were pre-incubated for 2 h in serum free media in the absence of M-CSF. After M-CSF addition, the cells were incubated for the indicated times and consequently harvested. The same protein amounts of cell lysates were subjected to SDS-PAGE followed by western blotting with antibodies against p-Erk, p-p38, p-Src, p-Akt and GAPDH. (b) BMMs from wild-type and ashen mice were pre-incubated for 2 h in serum free media in the absence of RANKL. After adding RANKL, the cells were incubated for the indicated times, consequently harvested. The same protein amounts of cell lysates were subjected to SDS-PAGE followed by western blotting with antibodies to p-Erk, p-p38, p-Akt and GAPDH.
Increased protein levels of c-fms (MCSF receptor) and RANK (RANKL receptor) in ashen OCLs
The mechanisms of enhanced signaling in ashen OCLs compared to wild-type OCLs were analyzed. First, the total protein levels of c-fms (M-CSF receptor) and RANK (RANKL receptor) in the precursor BMMs and mature OCLs were determined. We observed that the expression level of c-fms in ashen BMMs was higher than in wild-type BMMs (Fig. 5a). Densitometric analysis of these proteins also showed that the levels of c-fms in ashen BMMs or OCLs were significantly increased as compared to wild-type BMMs (Fig. 5b). However, RANK protein levels between wild-type and ashen BMMs were comparable (Fig. 5a, b). Interestingly, during differentiation of BMMs into mature OCLs, the expression levels of c-fms and RANK were significantly up-regulated in ashen OCLs compared to those in wild-type OCLs (Fig 5c, d).

Expression levels of RANK and c-fms in BMMs or OCLs from wild-type and ashen mice.
BMMs were prepared as per the method described in “Experimental Procedures”. The OCLs were further differentiated from BMMs cultured with RANKL (50 ng/ml) and M-CSF (30 ng/ml) for the 72 h. The same protein amounts of BMM-lysates (a) or OCL-lysates (c) were subjected to SDS-PAGE followed by western blotting with antibodies against RANK, c-fms and GAPDH. (b, d). Densitometric analysis for the quantification of each protein in the cell lysate of both cell types. The relative levels were defined as the chemiluminescence intensity per mm2 measured by LAS4000-mini. The data are represented as the mean ± S.D. of values from four independent experiments. *P < 0.05, **P < 0.01 for the indicated comparison. (e, f) Flow cytometric analysis of surface levels of c-fms (e) or RANK (f) in BMMs. The cells were stained for cell surface with specific antibody against c-fms or RANK and subsequently stained with a second antibody conjugated with PE. Data are representative of four independent experiments. (g) Down-regulation of RANK in BMMs from wild-type and ashen mice. Both the BMMs were treated with RANKL (50 ng/ml) and further incubated at 37°C for the indicated times. The cell lysates were subjected to SDS-PAGE followed by western blotting with antibodies against RANK and GAPDH. (h) Degradation of RANK by lysosomes in BMMs from wild-type and ashen mice. Both the BMMs were treated with RANKL (50 ng/ml) and further incubated at 37°C in the absence or presence of E-64 (10 micro g/ml) and pepstain (10 micro g/ml) for the indicated times. The cell lysates were subjected to SDS-PAGE followed by western blotting with antibodies against RANK and GAPDH.
We also investigated the surface expression levels of c-fms and RANK by flow cytometory (Fig. 5 e, f). The surface expression of c-fms was slightly higher in ashen BMMs than that observed in wild-type BMMs (Fig. 5e). However, the surface expression of RANK in ashen BMMs was comparable, but rather slightly lower than that observed in wild-type BMMs (Fig 5f). These results indicate that the enhanced signaling of c-fms in ashen cells is probably due to abnormal transport, accumulation and/or slightly increased surface expression levels.
Since we did not determine the mechanisms of the enhanced signaling of RANK in ashen BMMs, we further examined the possibility of down-regulation of RANK. Following stimulation with RANKL, the fate of the receptor was determined by western blotting (Fig. 5g). The cellular RANK in wild-type BMMs was gradually degraded for 4 h (Fig. 5g). In contrast, in ashen BMMs, the protein levels of RANK were rather increased at approximately 1–4 h (Fig. 5g). To evaluate the effects of lysosomal degradation of RANK, the protein levels of RANK after the ligand stimulation were determined by western blotting in the presence or absence of protease inhibitors such as E-64d, a cysteine protease inhibitor and pepstatin, an aspartic protease inhibitor (Fig. 5h). However, apparent differences were not observed between in wild-type and ashen BMMs (Fig. 5h). These results indicate that Rab27A deficiency caused the partially abnormal down-regulation or transport of RANK, but not abnormal receptor degradation by the lysosomes.
Taken together, we concluded that Rab27A deficiency in OCLs caused the enhanced M-CSF and RANKL signaling through abnormal transport of their respective receptors, c-fms and RANK and/or other adaptor proteins of these receptors.
Abnormal actin ring formation and altered localization of LAMP2 and CTSK in ashen OCLs
To determine whether Rab27A affects the transport of endosomes/lysosomes in OCLs or not, we analyzed actin-ring formation and subcellular localization of LAMP2 in wild-type and ashen OCLs on non-coated glass slip. We observed an ovary-shape and clear form in the actin-ring formation of wild-type OCLs, whereas ashen OCLs displayed an irregular shape and unclear form (Fig. 6a). The subcellular distribution of LAMP2 in wild-type OCLs was seen in various punctate structures located within the actin ring (Fig. 6a). However, the staining patterns showed two different localizations of LAMP2 in ashen OCLs: irregular-shaped punctate structures along the actin-ring distant from the nuclei (Fig. 6a, the upper panel) and those located predominantly around the nuclei (Fig. 6a, the lower panel). We also determined the subcellular localization of CTSK in wild-type and ashen OCLs. In wild-type OCLs, the staining of CTSK was detected in punctate structures within the central cytoplasm around the nuclei (Fig. 6b). However, in ashen OCLs, the subcellular localization of CTSK was predominantly present in the area circumscribed by the peripheral actin ring, although it was detected in various punctate structures around the multiple nuclei (Fig. 6b).

Immunofluorescence observation of actin ring formation and lysosomal proteins in OCLs from wild-type and ashen mice.
The cells on cover-glass were fixed, permeabilized with 0.3% Tween-20 in PBS and then allowed to react with phalloidin for F-actin (red) or antibodies for lysosomal proteins (green). After washing, the samples were incubated with a fluorescence-labeled secondary antibody and then were visualized by confocal laser microscopy. (a) On non-coated glass slip, LAMP-2 antibody. (b) On non-coated glass slip, CTSK antibody. (c) On vitronectin-coated glass slip, LAMP-2 antibody. (d) On vitronectin-coated glass slip, CTSK antibody. Bar: 50 μm.
To further observe on distribution of OCLs on dentin or bone slice, we performed alternative experiments on vitronectin-coated glass coverslips. Recently, Szewczyk et al. reported that the distribution of F-actin and lysosomal proteins in OCLs is very similar on bone slice and vitronectin-coated glass coverslips27. On vitronectin, ashen OCLs displayed more irregular shape of F-actin formation than wild-type OCLs (Fig. 6c, d), although the localizations of LAMP2 between wild-type and ashen OCLs were not so different (Fig. 6c, 6d). However, the localization of CTSK was exclusively around the nuclei in ashen OCLs, whereas that was detected in the central cytoplasm in wild-type OCLs. The results also indicate that the abnormal transport of LROs in ashen OCLs compared to wild-type OCLs.
Endosomal/lysosomal proteins in ashen OCLs are perturbed
The levels of various endosomal/lysosomal proteins in OCLs were determined by western blot analysis of wild-type and ashen OCL cell lysates. The expression level of LAMP-2 protein in ashen OCLs was lower than that in wild-type OCLs, although it was not statistically significant (Fig. 7). In contrast, the EEA1 and cathepsin D, (lysosomal aspartic protease), protein levels in ashen OCLs were higher than those in wild-type OCLs (Fig. 7). However, CTSK expression levels between wild-type and in ashen OCLs were similar (Fig. 7). In addition to lysosomal proteins, we found that expression of integrin β3, (OCL marker protein present in the plasma membrane) was significantly higher in ashen OCLs than that in wild-type OCLs (Fig. 7). On the other hand, the levels of Src, a cytosolic OCL marker protein, between wild-type and ashen OCLs were similar (Fig. 7). It should be noted that the protein expression of integrin β3 in ashen OCLs was highly increased compared with wild-type OCLs (Fig. 7), but the mRNA level of integrin β3 in ashen OCLs was comparable to wild-type OCLs (Fig. 3), suggesting that transport and/or degradation rate of integrin β3 between wild-type and ashen OCLs may be different. Thus, the expression levels of various endosomal/lysosomal proteins were perturbed in ashen OCLs compared to those in wild-type OCLs.

Lysosomal function in OCLs from wild-type and ashen mice.
(a) BMMs were prepared by the method described in “Experimental Procedures”. The OCLs were further differentiated BMMs that were cultured with RANKL (50 ng/ml) and M-CSF (10 ng/ml) for 6 days. The same protein amounts of OCL-lysates were subjected to SDS-PAGE followed by western blotting with antibodies to LAMP2, EEA1, cathepsin D (CTSD), CTSK, integrin β3, Src and GAPDH. (b) Densitometric analysis for the quantification of each protein in the cell lysate of both cell types. The relative levels were defined as the chemiluminescence intensity per mm2 measured by LAS4000-mini. The data are presented as the mean ± S.D. of values from four independent experiments. **P < 0.01 for the indicated comparison.
Impaired bone resorption activity in ashen OCLs
Finally, the bone resorption activity of OCLs was analyzed by a pit formation assay, using OCLs derived from wild-type and ashen mice. After comparing the resorption pit area, ashen OCLs exhibited a marked reduction in the resorbing activity, while wild-type OCLs had a moderate resorbing activity (Fig. 8a, b). Taken together, we concluded that the OCLs from ashen mice express an abnormal lysosome function.

Bone resorption activity of OCLs from wild-type and ashen mice.
(a)Photograph of the bone-resorption activity of OCLs from wild-type and ashen mice. (b) The bone resorption area was analyzed by using the BZ analyzer software, dynamic cell count system (Keyence). Data are presented as s the mean ± S.D. of three independent experiments. **P < 0.01 for the indicated comparisons.
Discussion
In this study, we have demonstrated the increase in Rab27A expression during OCL differentiation. Studies on siRNA knockdown in RAW-D cells or BMMs derived from ashen mice showed that Rab27A deficiency in OCLs is responsible for induction of multinucleation and giant cell formation. Stimulation with M-CSF or RANKL caused enhanced phosphorylation levels of Erk, p-38 and Src in ashen OCLs compared to wild-type OCLs. The ashen OCLs exhibited marked reduction in the bone resorbing activity and abnormal lysosome localization, indicating that Rab27A deficiency caused abnormal transport of LROs in the OCLs.
Rab27A-deficient OCLs were larger in size and multinucleated. This phenotype is probably due to the enhancement of several signaling pathways through cell surface receptors such as c-fms and RANK. Indeed, we observed increased phosphorylation levels of Erk, p-38 and Src signaling thorough the M-CSF c-fms complex and Erk, p-38 and Akt signaling through the RANKL-RANK complex in Rab27A-deficient OCLs. This phenotype is reminiscent of Vps35 deficiency in OCLs28. Vps35 is a critical essential component of the retromer protein complex that regulates transmembrane proteins from the endosomes to the Golgi apparatus. Vps35-deficient OCLs shows a larger cell size and are multinucleated due to enhanced RANK signaling28. In such a case, increased cell surface levels of RANK in Vps35-deficient OCLs causes enhanced RANK signaling, resulting in larger cell size and multinucleation. However, in case of Rab27A-deficient OCLs, the surface level of RANK was comparable to that of wild-type cells. On the other hand, the cell surface level of c-fms was increased in these cells compared to wild-type OCLs. One possible explanation that normal RANK surface level with enhanced RANKL signaling in Rab27-deficient OCLs is continuous remaining of RANK on endosomes. Alternatively, abnormal transport of adaptor proteins of RANK (e.g.TRAF6) in Rab27-deficient OCLs may cause the enhanced signaling.
Rab27A-deficient OCLs shows a markedly impaired bone resorption activity. Bone resorption is a complex process combining secretion of protons by the vacuolar H+-ATPase and various lysosomal enzymes and endocytosis/transcytosis of the degraded products via vesicle transport from apical to basolateral membrane29. Previous studies with various leukocytes have shown that Rab27A is involved in both exocytosis/secretion and endocytosis/phagocytosis30,31,32. Rab27A-deficient neutrophils33,34, eosinophils35, basophils36 all show impaired exocytosis. In contrast, receptor-mediated phagocytosis is enhanced in Rab27A-deficient macrophages. Rab27A-deficient dendritic cells show increased phagosome acidification and antigen degradation, causing a defect in antigen cross-presentation30. Thus, Rab27A mediated-bone resorption may be quite different mechanisms to its phagocytosis.
The LAMP2 distribution was observed in two different regions in ashen OCLs; the peripheral region along the actin-ring and the perinuclear region. Similarly, CTSK was predominantly detected in the similar peripheral region in ashen OCLs. Contrastingly, the distribution of LAMP2 and CTSK in wild-type OCLs was seen in various punctate structures within the actin ring, which is consistent with previous results37. The abnormal lysosomal protein distribution and severely impaired bone resorption activity in ashen OCLs suggests that Rab27A probably regulates cellular distribution and secretion of LROs in OCLs.
Rab27A is speculated to play a unique role in OCLs, since its depletion results in different phenotypes compared to that expressed during deletion of other Rab proteins in OCLs. Rab3, for example, is involved in regulated exocytosis in secretory cells such as neurons and neuroendocrine cells, whose characteristics are similar to those of Rab27A2. Rab3D is the Rab3 isoform predominantly expressed in OCLs. Although Rab3D-deficient OCLs exhibit an abnormal ruffled border, the localization of Rab3D in the post-TGN (trans-Golgi network) vesicles and not in the OCL lysosomes, suggests that Rab3D may regulate the biogenesis of a secretory compartment in the TGN38. Rab7 in OCLs is thought to be implicated in the formation of ruffled borders38. Rab7-gene knockdown experiments using antisense oligo-deoxynucleotides have revealed that Rab7 is required for polarization of ruffled border formation, as well as bone resorption by OCLs via transport of V-ATPase to the ruffled border membrane38. Rab13 is up-regulated during differentiation of human peripheral blood monocytic cells into OCLs39. Since Rab13 is localized in vesicles between the trans-Golgi network and basolateral membrane domain, it is not involved in endocytosis or transcytosis of bone degradation products39. Rab38 is involved in sorting of protein to the melanosome in melanocytes and phagosomal maturation in macrophages. However, OCLs derived from Rab38-deficient (Chocolate) mice show no phenotype, indicating redundant function(s) of other Rab protein(s)40. Considering the findings about other rab proteins in OCLs and our results, it is speculated that Rab27A may have redundant function of Rab38 in OCLs.
The role of Rab27A/B in osteoblasts has been reported. Suppression of Rab27A or RAb27B by RNA interference causes markedly reduced secretion of RANKL in murine osteoblastic cell line ST2 cells41. These results also suggest that Rab27deficiency in osteoblasts results in reduced OCL activation.
In conclusion, this study shows that Rab27A deficiency causes an abnormal transport of LROs in OCLs. Moreover, Rab27A deficiency shows the increased surface level of c-fms and the delayed down-regulation of RANK in ashen cells, resulting in enhanced signaling of M-CSF and RANKL.
Methods
Antibodies and reagents
Macrophage colony stimulating factor (M-CSF) was purchased from Kyowa Hakko Kogyo (Tokyo, Japan). Recombinant receptor activator of nuclear factor kappa-B ligand (RANKL) was prepared as described previously42. Antibodies (Abs) were purchased as follows: Rabbit polyclonal anti-GM130 (Cat. NO. PM061) and, Rabbit polyclonal anti-Calnexin (Cat.No. PM060) were from MBL (Nagoya, Japan). Rat monoclonal anti-LAMP2 (Cat. No. 1921-01) were from Southern Biotechnology Inc, Birmingham, AL, USA. Mouse monoclonal anti-Src (proto-oncogene tyrosine-protein kinase) (Cat. No. 05-184, Upstate Biotechnology, Lake Placid, NY, USA). Rabbit polyclonal anti-c-fms (colony-stimulating factor 1 receptor) (Cat. No. sc-692), Rabbit polyclonal anti-RANK (receptor activator of nuclear factor kappa-B) (Cat. No. sc-9072) and mouse monoclonal anti-NFATc1 (nuclear factor of activated T-cells, cytoplasmic 1) (Cat. No. sc-7294) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Abs specific for phospho-Erk1/2 (extracellular signal-regulated kinase) (Cat. No. 9101S, Thr202/Tyr204), phospho-Akt (protein kinase B) (Cat. No. 9271S, Ser473), phospho-p38 (Cat. No. 9211S, Thr180/Tyr182,) and phospho-Src (Cat No. 2105S, Tyr527) were from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal Anti-Rab27A/B from Immuno Biological Laboratories Ltd. (Takasaki, Japan). Rabbit polyclonal anti-cathepsin D or cathepsin K was prepared as described previously43,44. The Osteo Assay Plate was from Corning (Corning, NY, USA). All other reagents, including phenylmethylsulfonyl fluoride and the protease inhibitor cocktail, were from Sigma-Aldrich (Saint-Louis, MO, USA). Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 555 goat anti-rat IgG were from Molecular Probes/Invitrogen (Eugene, Oregon, USA).
Animals
The ash/ash (C3H/He background) and control C3H/He mice described previously were originally provided by N.A. Jenkins (National Cancer Institute, Frederick, Maryland, USA)45. All animals were maintained according to the guidelines offset by the Japanese Pharmacological Society. All animals, their handling and experiments were approved by the Animal Research Committee of the Graduate School of Biomedical Sciences, Nagasaki University.
Cell culture
Isolation of BMMs was performed according to a previously described method46. Briefly, marrow cells were cultured from mice femurs and tibias overnight in α-minimal essential medium (α-MEM) (Wako Pure Chemicals, Osaka, Japan) containing 10% fetal bovine serum (FBS), supplemented with 100 U/ml of penicillin and 100 μg/ml of streptomycin in the presence of M-CSF (10 ng/ml) at 37 °C in 5% CO2. The non-adherent cells were harvested; stroma-free bone marrow cells were cultured in the presence of 10 ng/ml of M-CSF. After 4 days, the non-adherent cells were washed out and the adherent cells were used as BMMs. The BMMs were replated and then further cultured with new medium containing M-CSF (10 ng/ml) and RANKL (50 ng/ml) for the times indicated.
mRNA expression profiling of OCL differentiation by Affymetrix Microarray
To identify the up-regulated genes during OCL differentiation, we utilized the Affymetrix microarray system (Affymetrix, Santa Clara, CA, USA). The data were obtained from BMMs cultured for 72 h on a plastic surface (rapid differentiation) and a dentin slice (slow differentiation) with M-CSF (30 ng/ml) and RANKL (50 μg/ml) were compared. Total RNA from the two types of BMMs were isolated with Trizol Reagent (Invitrogen, Carlsbad, USA). The samples were further purified using the RNeasy Mini Kit (QIAGEN, Tokyo, Japan) as per the manufacturer's instructions. The hybridized microarray gene chip arrays were analyzed according to the manufacturer's instructions and scanned by an Affymetrix Scanner.
Immunofluorescence microscopy
Immunocytochemistry was performed as described previously43. Briefly, the cells were grown on glass cover slips and fixed with 4.0% paraformaldehyde in PBS for 30 min at room temperature. The fixed cells were then washed with 50 mM NH4Cl in PBS for quenching and permeabilized with 0.2% Tween-20 in PBS for 10 min. The cells were incubated with 0.2% gelatin in PBS for 1 h and subsequently incubated overnight at 4°C with primary antibodies. After washing, the cells were stained by second antibodies such as Alexa Fluor 488 goat anti-rabbit IgG or Alexa Fluor 594 (Cell Signaling Technology, Danvers, MA, USA) goat anti-rabbit IgG. Actin cytoskeleton staining with Alexa 594-phalloidin (Invitrogen) or nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen Carlsbad, CA, USA) was performed. The samples were subjected to microscopy using a laser-scanning confocal imaging system (LSM510, 710 or 780 META; Carl Zeiss, AG, Jena, Germany).
Quantitative RT-PCR
Total RNA was isolated with Trizol Reagent and reverse transcription was performed using oligo(dT)15 primer (Promega) and Revertra Ace (Toyobo, Osaka, Japan). Quantitative real-time PCR was performed using a MX3005P QPCR system (Agilent Technology, La Jolla, CA, USA). cDNA was amplified using Brilliant III Ultra-Fast SYBR QPCR Master Mix (Agilent Technology) according to the manufacturer's instructions. The following primer sets were used: GAPDH forward, AAATGGTGAAGGTCGGTGTG; GAPDH reverse, TGAAGGGGTCGTTGATGG; Rab27A forward, TTCCTGCTTCTGTTCGACCT; Rab27A reverse, GGCAGCACTGGTTTCAAAAT; Rab27B forward, GATGGAGACTATGATTATCTG; Rab27B reverse, TCCATTGACAGTGTCCGGAACC; RANK forward, CTTGGACACCTGGAATGAAGAAG; RANK reverse, AGGGCCTTGCCTGCATC; c-fms forward, TTGGACTGGCTAGGGACATC; c-fms reverse, GGTTCAGACCAAGCGAGAAG; DC-STAMP forward, CTAGCTGGCTGGACTTCATCC; DC-STAMP reverse, TCATGCTGTCTAGGAGACCTC; OC-STAMP forward, TGGGCCTCCATATGACCTCGAGTAG; OC-STAMP reverse, TCAAAGGCTTGTAAATTGGAGGAGT; CTSK forward, CAGCTTCCCCAAGATGTGAT; CTSK reverse, AGCACCAACGAGAGGAGAAA; Integrin β3 forward, TGTGTGCCTGGTGCTCAGA; Integrin β3 reverse, AGCAGGTTCTCCTTCAGGTTACA; Src forward, AGAGTGCTGAGCGACCTGTGT; Src reverse, GCAGAGATGCTGCCTTGGTT; CTR forward, CGCATCCGCTTGAATGTG; CTR reverse, TCTGTCTTTCCCCAGGAAATGA.
Transient transfection of RAW-D cells
RAW-D cells were cultured in α-MEM supplemented with 10% FBS. All cells were maintained in a 70% confluent at 37°C in an incubator containing 5% CO2 and transiently transfected with pEGFP-C1-Rab27A using SuperFect™ Transfection Reagent (Quiagen, Germany) according to the manufacture's protocol.
Flow cytometry analysis
The cell suspension (2 × 106 cells/100 μl) was incubated on ice for 20 min with primary antibodies appropriately diluted with PBS containing 1% normal goat serum. Samples were preincubated with Fc Block (anti-mouse CD16/CD32 antibody) (BD Pharmigen) and subsequently with specific antibodies or control antibodies RANK (anti-mouse CD265-PE conjugated) or c-fms (anti-mouse CD115-PE conjugated) or isotype control (rat IgG2a k-PE conjugated) for 15 min on ice. Flow cytometric analyses were performed on a BD FACS Verse Cytometer.
TRAP staining method
The cells were fixed with 4% paraformaldehyde and stained for TRAP activity using a previously described method47 TRAP-positive cells with 3 or more nuclei were regarded as mature osteoclasts. Murine monocytic cell line RAW-D cells, were kindly provided by Prof. Toshio Kukita (Kyushu University, Japan) and cultured in α-MEM containing 10% FBS with RANKL (50 ng/ml)25 were used for this experiment.
Small interfering RNA (siRNA)
siRNA experiments in OCLs were performed as described previously48. The target sequence of murine Rab27A siRNA was: GGACUUAAUCAUCTAAGAGAAUGGAA (Rab27A siRNA). Briefly, RAW-D cells plated at 5 × 104 cells on 60-mm plates were cultured in the presence of RANKL in antibiotic-free media. The siRNA was transfected into RAW-D cells using Lipofectamine RNAiMAX™ transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. BLOCK-iT™ Alexa Fluor Red Fluorescent Oligo (Life Technologies, Carlsbad, CA, USA) was used to optimize the delivery of siRNA. The cells were incubated with 10 pmol of siRNA for 24 h. For TRAP staining, the cells were incubated for an additional 5 days.
Bone resorption assay
The bone-resorbing activity of OCLs was determined using the Osteo Assay Stripwell Plate for 5 days of culture. Images for cell area, bone resorption area and nuclei counting were taken with anal-in-one type fluorescence microscope BZ-9000 (Keyence). Deconvolution for the cell and bone resorption area was analyzed by using the BZ analyzer software, dynamic cell count system (Keyence).
RANK degradation assays
OCLs were grown in a 3.5 cm dish and incubated in α-MEM supplemented with 0.1% BSA lacking FBS for 2 h. Following this, they were incubated with 50 ng/ml of RANKL for the indicated period. The cells were harvested with a cell lysis buffer (50 mM Tris-HCl (pH 8.0), 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride and proteinase inhibitor cocktail).
Western blot analysis
BMMs were stimulated with or without RANKL in the presence of M-CSF for the indicated amount of time. Cells were rinsed twice with ice-cold PBS and lysed in a cell lysis buffer (50 mM Tris-HCl [pH 8.0], 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 1 mM PMSF and proteinase inhibitor cocktail). The protein concentration of each sample was measured with BCA Protein Assay Reagent (Thermo Pierce, Rockford, IL, USA) according to the manufacturer's instructions. Five micrograms of lysate protein was applied to each lane. Following SDS-PAGE, proteins were electroblotted onto a polyvinylidene difluoride membrane. The blots were blocked with 3% skim milk/TBS (Tris-buffered saline)-0.1%Tween for 1 h at room temperature, probed with primary antibodies overnight at 4°C. They were then washed and incubated with HRP-conjugated secondary antibodies. These were finally detected with ECL-prime (GE Healthcare Life Sciences, Little Chalfont, UK). The immunoreactive bands were analyzed using the LAS4000-mini (Fuji Photo Film, Tokyo, Japan).
Statistical analysis
Quantitative data are presented as means ± standard deviation (S.D.). The Tukey-Kramer method was used to identify differences between concentrations when ANOVA indicated that a significant difference (*P < 0.05 or **P < 0.01) existed.
References
Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10, 513–525, 10.1038/nrm2728 (2009).
Fukuda, M. Regulation of secretory vesicle traffic by Rab small GTPases. Cell Mol Life Sci 65, 2801–2813, 10.1007/s00018-008-8351-4 (2008).
Bizario, J. C. et al. Griscelli syndrome: characterization of a new mutation and rescue of T-cytotoxic activity by retroviral transfer of RAB27A gene. J Clin Immunol 24, 397–410, 10.1023/B:JOCI.0000029119 (2004).
Menasche, G. et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 25, 173–176, 10.1038/76024 (2000).
Johnson, J. L., Monfregola, J., Napolitano, G., Kiosses, W. B. & Catz, S. D. Vesicular trafficking through cortical actin during exocytosis is regulated by the Rab27a effector JFC1/Slp1 and the RhoA-GTPase-activating protein Gem-interacting protein. Mol Biol Cell 23, 1902–1916, 10.1091/mbc.E11-12-1001 (2012).
Dell'Angelica, E. C., Mullins, C., Caplan, S. & Bonifacino, J. S. Lysosome-related organelles. FASEB J 14, 1265–1278 (2000).
Marks, M. S., Heijnen, H. F. & Raposo, G. Lysosome-related organelles: unusual compartments become mainstream. Curr Opin Cell Biol 25, 495–505, 10.1016/j.ceb.2013.04.008 (2013).
Raposo, G. & Marks, M. S. The dark side of lysosome-related organelles: specialization of the endocytic pathway for melanosome biogenesis. Traffic 3, 237–248 (2002).
Booth, A. E., Seabra, M. C. & Hume, A. N. Rab27a and melanosomes: a model to investigate the membrane targeting of Rabs. Biochem Soc Trans 40, 1383–1388, 10.1042/BST20120200 (2012).
Haddad, E. K., Wu, X., Hammer, J. A., 3rd & Henkart, P. A. Defective granule exocytosis in Rab27a-deficient lymphocytes from Ashen mice. J Cell Biol 152, 835–842 (2001).
Novak, E. K. et al. The regulation of platelet-dense granules by Rab27a in the ashen mouse, a model of Hermansky-Pudlak and Griscelli syndromes, is granule-specific and dependent on genetic background. Blood 100, 128–135 (2002).
Baron, R., Neff, L., Louvard, D. & Courtoy, P. J. Cell-mediated extracellular acidification and bone resorption: evidence for a low pH in resorbing lacunae and localization of a 100-kD lysosomal membrane protein at the osteoclast ruffled border. J Cell Biol 101, 2210–2222 (1985).
Suda, T. et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev 20, 345–357 (1999).
Teitelbaum, S. L. & Ross, F. P. Genetic regulation of osteoclast development and function. Nat Rev Genet 4, 638–649, 10.1038/nrg1122 (2003).
Suda, T., Takahashi, N. & Martin, T. J. Modulation of osteoclast differentiation. Endocr Rev 13, 66–80 (1992).
Vaananen, H. K. & Horton, M. The osteoclast clear zone is a specialized cell-extracellular matrix adhesion structure. J Cell Sci 108 (Pt 8), 2729–2732 (1995).
Vaananen, H. K., Zhao, H., Mulari, M. & Halleen, J. M. The cell biology of osteoclast function. J Cell Sci 113 (Pt 3), 377–381 (2000).
Sobacchi, C., Schulz, A., Coxon, F. P., Villa, A. & Helfrich, M. H. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol 9, 522–536, 10.1038/nrendo.2013.137 (2013).
Lacombe, J., Karsenty, G. & Ferron, M. Regulation of lysosome biogenesis and functions in osteoclasts. Cell Cycle 12, 2744–2752, 10.4161/cc.25825 (2013).
Saftig, P. et al. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci U S A 95, 13453–13458 (1998).
Saftig, P. et al. Functions of cathepsin K in bone resorption. Lessons from cathepsin K deficient mice. Adv Exp Med Biol 477, 293–303, 10.1007/0-306-46826-3_32 (2000).
Lotinun, S. et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest 123, 666–681, 10.1172/jci64840 (2013).
Roberts, H. C. et al. Altered collagen in tartrate-resistant acid phosphatase (TRAP)-deficient mice: a role for TRAP in bone collagen metabolism. Calcif Tissue Int 80, 400–410, 10.1007/s00223-007-9032-2 (2007).
Halleen, J. M. et al. Intracellular fragmentation of bone resorption products by reactive oxygen species generated by osteoclastic tartrate-resistant acid phosphatase. J Biol Chem 274, 22907–22910 (1999).
Watanabe, T. et al. Direct stimulation of osteoclastogenesis by MIP-1alpha: evidence obtained from studies using RAW264 cell clone highly responsive to RANKL. J Endocrinol 180, 193–201 (2004).
Wilson, S. M. et al. A mutation in Rab27a causes the vesicle transport defects observed in ashen mice. Proc Natl Acad Sci U S A 97, 7933–7938 10.1073/pnas.140212797 (2000).
Szewczyk, K. A., Fuller, K. & Chambers, T. J. Distinctive subdomains in the resorbing surface of osteoclasts. PLoS One 8, e60285, 10.1371/journal.pone.0060285 (2013).
Xia, W. F. et al. Vps35 loss promotes hyperresorptive osteoclastogenesis and osteoporosis via sustained RANKL signaling. J Cell Biol 200, 821–837, 10.1083/jcb.201207154 (2013).
Oikawa, T., Kuroda, Y. & Matsuo, K. Regulation of osteoclasts by membrane-derived lipid mediators. Cell Mol Life Sci 70, 3341–3353, 10.1007/s00018-012-1238-4 (2013).
Jancic, C. et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol 9, 367–378, 10.1038/ncb1552 (2007).
Ejlerskov, P. et al. NADPH oxidase is internalized by clathrin-coated pits and localizes to a Rab27A/B GTPase-regulated secretory compartment in activated macrophages. J Biol Chem 287, 4835–4852, 10.1074/jbc.M111.293696 (2012).
Yokoyama, K. et al. Rab27a negatively regulates phagocytosis by prolongation of the actin-coating stage around phagosomes. J Biol Chem 286, 5375–5382, 10.1074/jbc.M110.171702 (2011).
Johnson, J. L. et al. Rab27a and Rab27b regulate neutrophil azurophilic granule exocytosis and NADPH oxidase activity by independent mechanisms. Traffic 11, 533–547, 10.1111/j.1600-0854.2009.01029.x (2010).
Johnson, J. L., Hong, H., Monfregola, J., Kiosses, W. B. & Catz, S. D. Munc13-4 restricts motility of Rab27a-expressing vesicles to facilitate lipopolysaccharide-induced priming of exocytosis in neutrophils. J Biol Chem 286, 5647–5656, 10.1074/jbc.M110.184762 (2011).
Kim, J. D. et al. An essential role for Rab27a GTPase in eosinophil exocytosis. J Leukoc Biol 94, 1265–1274, 10.1189/jlb.0812431 (2013).
Goishi, K., Mizuno, K., Nakanishi, H. & Sasaki, T. Involvement of Rab27 in antigen-induced histamine release from rat basophilic leukemia 2H3 cells. Biochem Biophys Res Commun 324, 294–301, 10.1016/j.bbrc.2004.09.050 (2004).
Toyomura, T. et al. From lysosomes to the plasma membrane: localization of vacuolar-type H+ -ATPase with the a3 isoform during osteoclast differentiation. J Biol Chem 278, 22023–22030, 10.1074/jbc.M302436200 (2003).
Zhao, H., Laitala-Leinonen, T., Parikka, V. & Vaananen, H. K. Downregulation of small GTPase Rab7 impairs osteoclast polarization and bone resorption. J Biol Chem 276, 39295–39302, 10.1074/jbc.M010999200 (2001).
Hirvonen, M. J. et al. Rab13 is upregulated during osteoclast differentiation and associates with small vesicles revealing polarized distribution in resorbing cells. J Histochem Cytochem 60, 537–549, 10.1369/0022155412448069 (2012).
Charles, J. F. et al. The collection of NFATc1-dependent transcripts in the osteoclast includes numerous genes non-essential to physiologic bone resorption. Bone 51, 902–912, 10.1016/j.bone.2012.08.113 (2012).
Kariya, Y. et al. Rab27a and Rab27b are involved in stimulation-dependent RANKL release from secretory lysosomes in osteoblastic cells. J Bone Miner Res 26, 689–703, 10.1002/jbmr.268 (2011).
Hu, J. P. et al. Berberine inhibits RANKL-induced osteoclast formation and survival through suppressing the NF-kappaB and Akt pathways. Eur J Pharmacol 580, 70–79, 10.1016/j.ejphar.2007.11.013 (2008).
Yanagawa, M. et al. Cathepsin E deficiency induces a novel form of lysosomal storage disorder showing the accumulation of lysosomal membrane sialoglycoproteins and the elevation of lysosomal pH in macrophages. J Biol Chem 282, 1851–1862, 10.1074/jbc.M604143200 (2007).
Kamiya, T. et al. Fluorescence microscopic demonstration of cathepsin K activity as the major lysosomal cysteine proteinase in osteoclasts. J Biochem 123, 752–759 (1998).
Kasai, K. et al. Rab27a mediates the tight docking of insulin granules onto the plasma membrane during glucose stimulation. J Clin Invest 115, 388–396, 10.1172/JCI22955 (2005).
Sakai, E. et al. Suppression of RANKL-dependent heme oxygenase-1 is required for high mobility group box 1 release and osteoclastogenesis. J Cell Biochem 113, 486–498, 10.1002/jcb.23372 (2012).
Hotokezaka, H. et al. U0126 and PD98059, specific inhibitors of MEK, accelerate differentiation of RAW264.7 cells into osteoclast-like cells. J Biol Chem 277, 47366–47372, 10.1074/jbc.M208284200 (2002).
Sakai, E. et al. Fisetin inhibits osteoclastogenesis through prevention of RANKL-induced ROS production by Nrf2-mediated up-regulation of phase II antioxidant enzymes. J Pharmacol Sci 121, 288–298, 10.1254/jphs.12243FP (2013).
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan to T.T. and for Research Fellow of Japan Society for the Promotion of Science to M.S.-S. We thank Dr. K. Nishishita for preparation of RANKL and Dr. Y. Fukuma and Mr. Y. Yamaguchi for technical assistance.
Author information
Authors and Affiliations
Contributions
M.S.-S. and E.S. performed the experiments. M.S.-S., E.S. and K.O. analyzed the data. M.F., T.I. and N.Y. contributed materials and provided fruitful discussion. T.T. designated and wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shimada-Sugawara, M., Sakai, E., Okamoto, K. et al. Rab27A Regulates Transport of Cell Surface Receptors Modulating Multinucleation and Lysosome-Related Organelles in Osteoclasts. Sci Rep 5, 9620 (2015). https://doi.org/10.1038/srep09620
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09620
This article is cited by
-
Abr, a Rho-regulating protein, modulates osteoclastogenesis by enhancing lamellipodia formation by interacting with poly(ADP-ribose) glycohydrolase
Molecular Biology Reports (2023)
-
Lack of Rab27a attenuates foam cell formation and macrophage inflammation in uremic apolipoprotein E knockout mice
Journal of Molecular Histology (2023)
-
Expression and localisation of Rab44 in immune-related cells change during cell differentiation and stimulation
Scientific Reports (2020)
-
KBTBD11, a novel BTB-Kelch protein, is a negative regulator of osteoclastogenesis through controlling Cullin3-mediated ubiquitination of NFATc1
Scientific Reports (2019)
-
Essential Role of the a3 Isoform of V-ATPase in Secretory Lysosome Trafficking via Rab7 Recruitment
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
