
Abstract
It is unclear whether N-acetylaspartate (NAA) depletions documented in schizophrenia patients might be due to the disease progression or medications. Here we investigated longitudinal NAA changes in drug-naïve first-episode patients (FEP) who are relatively free from chronicity. Forty-two drug-naïve FEP and 38 controls were enrolled in this study to explore the effect of 8-week risperidone monotherapy on NAA. All spectra were obtained from the medial prefrontal cortex (MPFC) on a 3.0 T MRI and analyzed with LCModel. At baseline, patients presented no significant differences in NAA (P = 0.084) or NAA/Cr + Pcr (P = 0.500) compared to controls; NAA levels were negatively correlated with PANSS total scores (P = 0.001) and WCST-PE (P = 0.041). After treatment, patients demonstrated significant reductions of NAA (P < 0.001) and NAA/Cr + Pcr (P < 0.001) and significant improvement in PANSS-P (P < 0.001) and PANSS-G (P < 0.001) symptoms. We detected no significant correlations between NAA alterations and PANSS-P (P = 0.679) or PANSS-G (P = 0.668) symptom changes; nor did NAA/Cr + Pcr changes with alterations in PANSS-P (P = 0.677) and PANSS-G (P = 0.616). This is the first evidence that short-term risperidone treatment induces an acute reduction of MPFC NAA during the early phase of schizophrenia, which may be a previously unavailable biomarker to indicate risperidone with a similar pharmacological mechanism, although the functional significance is still unclear.
Similar content being viewed by others
Introduction
Schizophrenia (SZ) is a complicated mental disorder and characterized by a cluster of symptoms, such as positive and negative symptoms, cognitive deficits and so on. Recently, abnormalities of brain metabolites, such as bioenergetic molecules or neurotransmitters, have been an alternative perspective to explore the pathophysiology of SZ. The amino acid of N-acetylaspartate (NAA) has been widely investigated in many clinical studies1,2,3, as two primary factors bring it to the attention of the neuroscience. First is its exclusive localization in neurons (not in glial cells), which makes it one of the reliable markers of neuronal number or function4. The second is its prominent proton signal in proton magnetic resonance spectroscopy (1H-MRS), which makes it one of the most accurate and reliable markers for 1H-MRS researches5.
Cross-sectional 1H-MRS studies have shown consistent findings that chronic SZ patients have significant NAA depletion in the prefrontal cortex (PFC)6,7, while several other studies didnot observe NAA anomalies in the PFC in the early phase of SZ8,9,10. In one recent study, Natsubori et al11 investigated the subjects at three different stages of illness including chronic patients, FEP and ultrahigh risk populations and found that significant reduction of NAA in the medial PFC (MPFC) exists specifically in chronic subjects but not in FEP or ultrahigh risk ones. Consistently, Ohrmann et al12 suggested that chronic patients but not FEP have NAA declines in the PFC. These may reflect the progressive alterations of neurometabolites in SZ13. However, besides the disease itself, many other factors including treatment effects may also affect the NAA deficits observed in chronic SZ through cross-sectional studies.
In the pharmacological properties of neuroleptics, D2 receptor antagonism plays a key role in alleviating patients' positive symptoms, although, the relative success is limited by the lack of efficacy for negative symptoms and cognitive impairments14. The severity of negative symptoms and cognitive impairments of SZ patients is related to the reduction of D1 receptors in the PFC15. Unfortunately, blockade of D2 receptor16 or antipsychotics exposure17 can induce down-regulation of D1 receptors in the PFC. Additionally, it has been speculated that the mitochondria effects of antipsychotics reported broadly18,19,20 are possibly regulated by downstream effects following blockade of relative dopamine receptors in the cortex21. Moreover, D1 receptor agonists can partially prevent neuroleptic-induced deleterious influence on mitochondrial function22. Importantly, NAA is exclusively synthesized in neuronal mitochondria and mitochondrial impairments can rapidly affect NAA production23.
Longitudinal designs can partly overcome the unresolved problem in cross-sectional studies and relatively distinguish between the progressive pathogenesis and the treatment effects. However, recently, longitudinal 1H-MRS studies investigating the influence of neuroleptics on central NAA have yielded inconsistent findings8,24,25. Most studies have used chronically treated patients1,25,26 or minimally treated patients8,24 and the few studies of drug-naïve patients2 are limited by medication with heterogeneous drugs. Therefore, the confounders of medications and chronicity can not be completely excluded and the effect of antipsychotics on NAA still has to be explored. First-episode and drug-naïve SZ patients are in the very early phase of illness when relatively ‘free’ from prior treatment and chronicity. To date, there is still lack of longitudinal studies with large samples of drug-naïve first-episode patients (FEP) to investigate NAA alterations after short-term monotherapy.
Therefore, we performed this longitudinal study of 1H-MRS in a relatively large cohort of drug-naïve FEP to explore the NAA changes after 8-week treatment withrisperidone monotherapy. We selected MPFC as the voxel of interest. We test the hypothesis that first-episode SZ patients would have alterations in NAA levels after treatment. We also conducted an exploratory research to analyse whether NAA changes would be relevant to alterations in clinical symptoms and cognitive function.
Results
Demographic and clinical characteristics between cases and controls at Baseline
Forty-two patients met the criteria of drug-naïve first-episode SZ (duration of illness = 8.38 ± 2.61 months). As shown in table 1, patient and control groups were well matched in age, gender, education, handedness and alcohol and tobaco usage (Ps > 0.05). However, significant lower performance in WCST categories (t78 = 2.718, P = 0.008) and WCST persererative errors (WCST-PE) (t78 = 9.041, P < 0.001) were observed in case group.
Longitudinal comparisons of clinical symptoms and cognitive function between baseline and week 8
As shown in table 1, we observed significant decreases in PANSS total scores (PANSS-T) (t41 = 13.19, P < 0.001), PANSS positive symptom scores (PANSS-P) (t41 = 14.58, P < 0.001) and PANSS general psychopathological symptom scores (PANSS-G) (t41 = 13.37, P < 0.001), while no significant changes in PANSS-N (t41 = 1.42, P = 0.163) after treatment. We detected no significant changes in WCST categories (t41 = 1.46, P = 0.151) and WCST-PE (t41 = 1.72, P = 0.093) after treatment. Therefore changes of PANSS-N and WCST performance were not further calculated with the Pearson correlation analysis.
Quality of 1H-MRS spectra
The mean ± SD measures of FWHM from LCModel were 0.065 ± 0.017 ppm for patients at baseline, 0.060 ± 0.018 ppm for patients at 8-week follow-up and 0.067 ± 0.018 ppm for controls. The mean ± SD measures of S/N from LCModel were 21.36 ± 2.35 for patients at baseline, 20.03 ± 2.36 for patients at 8-week follow-up and 20.58 ± 3.23 for controls. There were no significant differences in both of the two measures in the three groups: F2 = 1.492, P = 0.229 for FWHM; and F2 = 2.510, P = 0.086 for S/N. The range of Cramer–Rao minimum variance of NAA was from 1% to 10% in the three groups.
Comparison of baseline NAA and NAA/Cr + Pcr between cases and controls and longitudinal changes following treatment
At baseline, no significant differences were observed between patients and controls in terms of NAA levels (t78 = 1.751, P = 0.084, figure 1a), NAA/Cr + Pcr (t78 = 0.677, P = 0.500, figure 1b) and Cr + Pcr (t78 = 0.541, P = 0.590). After 8-week treatment, there were significant declines in NAA levels (t37 = 5.187, P < 0.001, figure 1c) and NAA/Cr + Pcr (t37 = 4.097, P < 0.001, figure 1d) in patients, but no significant alterations of Cr + Pcr levels (t37 = 1.210, P = 0.234). Moreover, responding patients showed no differences in baseline NAA, NAA/Cr + Pcr or Cr + Pcr (Ps > 0.05) relative to non-responding patients (details shown in Table S1). After treatment, the changes of NAA or NAA/Cr + Pcr demonstrated no significant differences between responding and non-responding patients (Ps > 0.05) (Table S2).

Medial prefrontal cortex NAA levels and NAA/Cr + Pcr ratio of each subject in schizophrenia patients and controls at baseline and after 8-week treatment.
(a) At baseline patients had no significant difference in NAA levels compared to controls (t78 = 1.75, P = 0.084, independent-samples t tests). (b) At baseline patients had no significant difference in NAA/Cr + Pcr compared to controls (t78 = 0.68, P = 0.500 independent-samples t tests). (c) Patients had significant reductions in NAA levels following 8-week treatment (t37 = 6.98, P < 0.001, paired-samples t tests). (d) Patients had a significant reduction in NAA/Cr after 8-week treatment (t37 = 5.38, P < 0.001, paired-samples t tests). *statistical levels of significance.
Correlations of Baseline NAA or NAA/Cr + Pcr with Clinical and Demographic Characteristics in Patients
At baseline, we noted significantly negative correlations of NAA levels with PANSS-total scores (γ = −0.502, P = 0.001, Figure 2a) and WCST-PE (γ = −0.316, P = 0.041, Figure 2b). There were no significant correlations between NAA levels and PANSS-P, PANSS-G or PANSS-N (Ps > 0.05). NAA/Cr + Pcr was not related to PANSS-T, PANSS-P, PANSS-G, PANSS-N or WCST-PE (Ps > 0.05). Moreover, NAA levels or NAA/Cr + Pcr didnot show significant correlatioins with age, education and illness duration prior to treatment (Ps > 0.05). Statistical analysis also indicated that NAA levels or NAA/Cr + Pcr were not influenced by gender (t40 = −0.156, P = 0.876; t40 = 0.895, P = 0.376, respectively), tobaco use (t40 = −0.092, P = 0.927; t40 = −1.132, P = 0.264, respectively), alcohol use (t40 = −0.413, P = 0.681; t40 = −0.646, P = 0.522, respectively) and family history (t40 = 1.746, P = 0.088; t40 = 1.195, P = 0.239, respectively). We showed the distribution of the NAA levels cross education and age in both patients and controls in Figure S1.

Correlations of NAA levels with clinical symptoms or cognitive function in patients at baseline.
(a) NAA levels were negatively related to PANSS total scores in 42 patients at baseline (γ = −0.502, P = 0.001). (b) NAA levels were negatively correlated with Wisconsin Card Sorting Test– perseveration errors in 42 patients at baseline (γ = −0.316, P = 0.041). Abbreviations: PANSS–T, PANSS total scores; WCST–PE, Wisconsin Card Sorting Test–perseveration errors.
Correlations of NAA alterations with symptom changes after treatment
After 8-week treatment, we observed no significant correlations between changes in NAA levels and alterations in PANSS-T (PANSS reductive ratio) (γ = −0.289, P = 0.088), PANSS-P (γ = −0.238, P = 0.162) and PANSS-G (γ = −0.278, P = 0.101). Nor did changes in NAA/Cr + Pcr with alterations in PANSS-T (γ = −0.255, P = 0.134), PANSS-P (γ = −0.145, P = 0.400) and PANSS-G (γ = −0.291, P = 0.094). In addition, acute alterations of NAA levels and NAA/Cr + Pcr following drug treatment were not influenced by age (γ = 0.145, P = 0.384; γ = −0.230, P = 0.165, respectively), education (γ = −0.072, P = 0.669; γ = 0.045, P = 0.788, respectively), illness duration prior to treatment (γ = 0.179, P = 0.283; γ = 0.093, P = 0.580, respectively), gender (t36 = −0.332, P = 0.742; t36 = −0.549, P = 0.586, respectively), tobaco use (t36 = 0.453, P = 0.662; t36 = 1.137, P = 0.263, respectively) and alcohol use (t36 = 1.692, P = 0.099; t36 = 1.313, P = 0.198, respectively).
Discussion
The key finding of our present study was that there was an acute decline of MPFC NAA and NAA/Cr + Pcr after 8-week risperidone exposure in drug-naïve FEP. To our knowledge, this study provides the first evidence that short-term risperidone treatment induces reductions of NAA in the MPFC in the early phase of SZ. Importantly, we found no significant correlations of the baseline NAA and NAA/Cr + Pcr with the illness duration and changes of NAA levels and the NAA/Cr following treatment were not affected by the illness duration. These results indicated that the acute NAA decline was more likely to be the result of medications rather than the disease progression. Moreover, patients have no significant differences in the baseline NAA and NAA/Cr + Pcr compared to controls and they have experienced psychotic symptoms for 8.38 ± 2.61 months. Therefore, the treatment effect is more reasonable to explain for the acute NAA depletion.
Recently, Lieberman et al13 completed the first meta-analysis of 1H-MRS findings, comparing different brain regions in individuals from prodrome to chronic SZ (97 studies: 2477 chronic patients, 923 FEP, 392 individuals at high risk of SZ and 3103 controls). Concurring with our present results, for FEP in the frontal lobe, illness duration presents no significant influence on NAA; while significant effects of antipsychotic medications on NAA were found (lower NAA levels with longer duration of medications). Their findings supported the hypothesis that the antipsychotic treatment rather than the illness progression is associated with the decreased frontal NAA in FEP.
Combining with the mounting available evidence23,27,28, the acute NAA decline after medications in this study may be a marker of reversible neuronal mitochondrial dysfunction, rather than permanent apoptosis. The NAA depletion in our study would be better understood in the context of pharmacologic properties of risperidone. Although risperidone has low affinity for D1 receptors29, exposure to risperidone can induce approximately two-thirds reduction of D1 receptors expression in primates PFC17. Furthermore, it has reported that D1 rather than D2 receptor agonists, can partially prevent neuroleptic-induced deleterious influence on the function of neuronal mitochondria22. Thus, it is possible that the adverse effect of risperidone on NAA depletions in the MPFC presented in this study may caused by, at least partly, a drug-related D1 receptors down-regulation in the MPFC. Additionally, unlike haloperidol, risperidone firstly inhibites neuronal dyfunction in the frontal cortex, but not in the extrapyramidal regions21, which consists with our present findings that the MPFC NAA decline occured in the absence of extrapyramidal adverse effects.
In this study, patients were significantly benefited from medications in terms of improved positive symptoms. This may be due to the fact that D2 receptor antagonism of risperidone plays a key role in alleviating patients' positive symptoms. However, their negative symptoms and poor performance of WCST were not improved. Positron emission tomography (PET) study demonstrated that reductions of PFC D1 receptor in drug-free or drug-naïve SZ patients are significantly related to their negative symptoms and poor performance of WCST15. In this case, risperidone-elicited down-regulation of D1 receptor might further exacerbate patients' baseline negative symptoms and poor performance of WCST, which appears to contradict our findings of no changes in negative symptoms and WCST performance. However, we still can't fully rule out the possibility that patients' negative symptoms and impaired cognitive function would be further aggravated if medications had been prolonged and that the aggravation might be related to decreased NAA. This possibility is supported by the important findings in the meta-analysis of Lieberman et al13 that lower NAA is related to higher negative symptom scores in chronically treated patients. Additionally, since the risperidone effect on D1 receptor down-expression may be not related to alterations of positive symptoms, in this study the NAA changes were not related to the alterations of positive symptoms. Therefore, NAA depletions may be secondary to treatment-related down-regulation of D1 receptor in the MPFC.
Our findings of early NAA defecits after risperidone medications stand in contrast to the available clinical 1H-MRS researches that have reported a modest increase1,2,26,30,31 or no changes1,24 in NAA levels or NAA/Cr after treatment with antipsychotics32, including risperidone. The discrepancy may be due to several factors. First, sample variations, such as sample sizes, ethnicity, symptoms severity, length of untreated psychotic symptoms and so on, may contribute to inconsistent findings. Second, according to the speculation that the risperidone-related down-regulation of D1 receptors in the MPFC may have detrimental effects on neuronal NAA, different antipsychotics may bring discrepant effects on metabolite levels in the same brain area due to different pharmacological properties. Therefore it seems appropriate for future studies to investigate the effect of each drug separately in large samples of drug-naïve patients. Third and perhaps most important, different lengths of follow-up may lead to an artificial discrepancy and heterogeneity. If NAA levels are influenced independently by antipsychotics but not the disease progression (although to date it is difficult to absolutely exclude the effects of illness itself), we speculate that there appears to be a nonlinear correlation between the antipsychotic exposure and NAA levels. If drug-induced NAA alterations occur very early (eg. 8–12 weeks), shorter or longer follow-ups in subjects may obscure the antipsychotic effects on NAA levels, namely, patients followed longer may have less or no NAA reductions because the NAA decline may occur earlier.
Another finding in this study is that the baseline NAA level was negatively correlated with PANSS-T and WCST-PE, namely, higher NAA levels may be beneficial for patients to experience less serious symptoms and cognitive impairment, which consists with broad studies12,25,31,32,33. However, in this case, decreased NAA levels after treatment would have been related to exacerbated clinical presentations, but in this study patients' PANSS-T symptoms improved and their poor performance of WCST remained stable. Furthermore, the alterations of NAA or NAA/Cr + Pcr after treatment showed no significant differences between responding and non-responding patients. On integrating our present results with that of Szulc et al25, Ertugrul et al31 and Bustillo et al8, the evidence suggests that the marked reduction of the MPFC NAA following the risperidone treatment may be a previously unavailable biomarker to indicate the antipsychotic drugs with a similar pharmacological mechanism, although the functional significance is still unclear.
Additionally, patients had no significant differences in baseline NAA and NAA/Cr + Pcr in the MPFC compared to controls. Cross-sectional 1H-MRS studies have broadly found NAA reductions in the thalamus, hippocampus and dorsolateral PFC primarily in chronic patients, but also in FEP, however, changes of MPFC NAA in SZ yet seem to be inconclusive6. Whether SZ patients exist abnormalities of MPFC NAA should be further explored.
Certain limitations of our present study need considerations. First, even though drug-naïve FEP are in the very early phase of illness when “free” from chronicity relatively, the influence of the potential progressive characteristic of SZ on NAA depletions can not be fully excluded. Second, due to the absence of data about the brain metabolite alterations after medication withdrawal or the relative parallel research design of neuroprotective therapies, we can't absolutely clarify whether the early acute NAA defecit is a marker of neuron apaptosis or dysfunction6. Third, although, to date, this is the largest longitudinal study in first-episode drug-naïve patients given risperidone monotherapy, the sample size perhaps still lacks power to completely eliminate the possibility that the NAA change is an acute phe-random and not systematically affect one group or the other.
In conclusion, our present findings demonstrated a possible evidence that risperidone monotherapy induces significant NAA depletions in the MPFC in the early phase of SZ, although a potential effect of the disease progression still cannot be excluded completely. Our findings may shed light on the nature of detrimental effect of risperidone on neuronal function during the early phase of SZ. We underscore the need for longer follow-up to examine whether negative symptoms and cognitive function of SZ patients would be further exacerbated and whether the exacerbation would be related to NAA depletions following prolonged medications. Additionally, our observations may indicate that MPFC NAA is a biomarker of baseline symptom severity and cognitive function.
Methods
Participants
Forty-two in-patients were recruited from August 2012 to December 2013 at the Second Affiliated Hospital, Xinxiang Medical University, Xinxiang, Henan, China. All patients were drug-naïve and were experiencing their first-episode psychosis when enrolled to this study. They were diagnosed by one of two trained psychiatrists according to the Structured Clinical Interview for DSM-IV-TR (Diagnostic and Statistical Manual of Mental Disorders, forth edition, American Psychiatric Association, 2000), patient version (SCID-I/P) and all patients met the diagnostic criteria for SZ. Thirty-five patients had been diagnosed as paranoid schizophrenia, while 7 patients undifferentiated schizophrenia. The patients had an onset age from 18 to 45 years and had less than 1 year duration of disease. Thirty-eight gender-, age- and education-matched healthy controls were recruited by advertisement. All controls were screened for lifetime absence of psychiatric diseases by using the SCID, Non-Patient Edition and were confirmed to be without a family history of psychosis in their first-degree relatives. All participants were Chinese Han people and right-handed. Participants with a history of neurological illness, physical disorders or substance abuse were excluded from this study.
This study was approved by the Ethics Committee of the Second Xiangya Hospital of Central South University (No. S088, 2012) and the cooperated hospital, the Second Affiliated Hospital of Xinxiang Medical University. This study was carried out in accordance with the Declaration of Helsinki. Signed informed consent forms were obtained from all participants. All patients were aware that they were entitled to withdraw from this study at any time without jeopardizing their clinical treatment. Four patients did withdraw at the follow-up 1H-MRS scanning, but they have completed clinical assessment.
Medication
All patients took risperidone monotherapy for 8 weeks. Concomitant treatment with mood stabilizers or antidepressants was not permitted. The dose titration was according to the clinical status of patients. Compliance with risperidone was monitored by clinical interviews once a week. No serious side effects were reported during the treatment.
1H-MRS scanning procedure
All patients had 1H-MRS scans within 24 h after included in this study when they were free from drugs and scans were scheduled again for patients after 8-week treatment, while controls only had baseline scans. All examinations were completed on a 3.0 T magnetic resonance imager (Siemens, Verio, Germany) using a 16-channel head coil at the Magnetic Imaging Centre of the Second Affiliated Hospital of Xinxiang Medical University. Participants were fitted with earplugs and foam pads to reduce scanner noise and limit head motion. T1-weighted anatomical images were obtained with 3-dimensional rapid acquisition with gradient echo for voxel tissue segmentation (TR 1900 ms; TE 2.52 ms; FOV 250 × 250 mm; flip angle 9°; slice thickness 1 mm; gap 0 mm; NEX 1; slices 176). 1H-MRS spectra were acquired using the standard PRESS sequence (svs_se, TR 8s, TE 80 ms, NEX 128). A 20 × 20 × 25 mm voxel of interesting (VOI) was prescribed to include mostly gray matter in the medial prefrontal cortex using coronal, sagittal and transverse images (Figure 3). The unsuppressed water spectra were also scanned in the same voxel. The priliminary quality detection of spectra was performed with the spectroscopic processing of Siemens Spectroscopy application (Syngo B17, Germany). Spectra with apparent baseline drift were excluded and scanning would be repeated again after patients' consent was obtained.

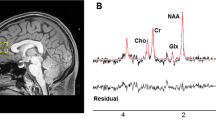
Spectroscopic voxel placement and proton magnetic resonance (1H-MRS) spectra.
(a) Voxel placement in medial prefrontal cortex (MPFC). (b) Sample 1H-MRS spectra acquired from MPFC as fitted by LCModel (provencher, 1993).
NAA was quantified using LCModel version 6.3-1B (LCMODEL Inc. CA) at the Second Affiliated Hospital, Shantou University Medical College, Shantou, Guangdong. The cerebrospinal fluid (CSF) content in the MPFC was served as the internal standard for calculation of absolute concentration of NAA. NAA scaled to creatine plus phosphocreatine (NAA/Cr + Pcr) was also selected as an important value reported by LCModel, as it is also a helpful measure of neuronal integrity34. To ensure data quality and avoid bias, any spectra that met one or more of these criteria was discarded: (1) the Cramer–Rao minimum variance > 15%; (2) NAA FWHM < 0.1; and (3) the signal-to-noise ratio (S/N) > 10. More details are available in the LCModel manual (http://s-provencher.com/pages/lcm-manual.shtml).
Clinical assessments
Patients (both at baseline and week 8) and controls (only at baseline) completed computerized 128-card version of the Wisconsin Card Sorting Test (WCST), which can evaluate subjects' executive function of frontal cortex, including perseveration, flexibility and abstract problem solving strategies35. Both at baseline and week 8, symptoms severity of patients was measured by using the 30-item Positive and Negative Syndrome Scale (PANSS)36.
Statistical analyses
Statistical analyses were completed using SPSS 13.0 (SPSS Inc., USA). Group differences of demographic data, metabolites levels and WCST scores were performed using independent-samples t tests or χ2 square test. Clinical symptom scores and WCST performance of patients between the two time points (baseline and week 8) were performed using paired-samples t tests. Correlations between alterations in brain metabolites and changes in clinical symptoms or WCST scores were further calculated with the partial correlation analysis (with baseline symptom scores or metabolite levels as unconcerned confounding factors) only if there was a significant variation between the two time points. The significance level was set with a threshold of P < 0.05.
References
Bertolino, A. et al. The effect of treatment with antipsychotic drugs on brain N-acetylaspartate measures in patients with schizophrenia. Biol. Psychiatry 49, 39–46 (2001).
Fannon, D. et al. Selective deficit of hippocampal N-acetylaspartate in antipsychotic-naive patients with schizophrenia. Biol. Psychiatry 54, 587–598 (2003).
Bustillo, J. et al. 1H-MRS at 4 tesla in minimally treated early schizophrenia. Mol. psychiatry 15, 629–636 (2010).
Lutkenhoff, E. S. et al. Proton MRS in twin pairs discordant for schizophrenia. Mol. Psychiatry 15, 308–318 (2010).
Luyten, P. R. & Den Hollander, J. A. Observation of metabolites in the human brain by MR spectroscopy. Radiology 161, 795–798 (1986).
Schwerk, A. et al. Metabolic alterations associated with schizophrenia: a critical evaluation of proton magnetic resonance spectroscopy studies. J. Neurochem. 128, 1–87 (2014).
Molina, V. et al. Dorsolateral prefrontal N-acetyl-aspartate concentration in male patients with chronic schizophrenia and with chronic bipolar disorder. Eur. Psychiatry 22, 505–512 (2007).
Bustillo, J. R. et al. Longitudinal follow-up of neurochemical changes during the first year of antipsychotic treatment in schizophrenia patients with minimal previous medication exposure. Schizophr. Res. 58, 313–321 (2002).
Goto, N. et al. Comparison of brain N-acetylaspartate levels and serum brain-derived neurotrophic factor (BDNF) levels between patients with first-episode schizophrenia psychosis and healthy controls. Eur. Psychiatry 26, 57–63 (2011).
Galinska, B. et al. Duration of untreated psychosis and proton magnetic resonance spectroscopy (1H-MRS) findings in first-episode schizophrenia. Med. Sci. Monit. 15, CR82–88 (2009).
Natsubori, T. et al. Reduced Frontal Glutamate + Glutamine and N-Acetylaspartate Levels in Patients With Chronic Schizophrenia but not in Those at Clinical High Risk for Psychosis or With First-Episode Schizophrenia. Schizophr. Bull 40 (2013).
Ohrmann, P. et al. Cognitive impairment and in vivo metabolites in first-episode neuroleptic-naive and chronic medicated schizophrenic patients: a proton magnetic resonance spectroscopy study. J. Psychiatr. Res. 41, 625–634 (2007).
Brugger, S. et al. Proton magnetic resonance spectroscopy and illness stage in schizophrenia--a systematic review and meta-analysis. Biol. Psychiatry 69, 495–503 (2011).
Miyamoto, S. et al. Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol. Psychiatry 17, 1206–1227 (2012).
Okubo, Y. et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 385, 634–636 (1997).
Castner, S. A. et al. Reversal of antipsychotic-induced working memory deficits by short-term dopamine D1 receptor stimulation. Science 287, 2020–2022 (2000).
Lidow, M. S. et al. Down-regulation of the D1 and D5 dopamine receptors in the primate prefrontal cortex by chronic treatment with antipsychotic drugs. J. Pharmacol. Exp. Ther 281, 597–603 (1997).
Burkhardt, C. et al. Neuroleptic medications inhibit complex I of the electron transport chain. Ann. Neurol 33, 512–517 (1993).
Balijepalli, S. et al. Inhibition of mitochondrial complex I by haloperidol: the role of thiol oxidation. Neuropharmacology 38, 567–577 (1999).
Casademont, J. et al. Neuroleptic treatment effect on mitochondrial electron transport chain: peripheral blood mononuclear cells analysis in psychotic patients. J. Clin. Psychopharmacol 27, 284–288 (2007).
Balijepalli, S. et al. Protein thiol oxidation by haloperidol results in inhibition of mitochondrial complex I in brain regions: comparison with atypical antipsychotics. Neurochem. Int 38, 425–435 (2001).
Barrientos, A. et al. Biochemical and molecular effects of chronic haloperidol administration on brain and muscle mitochondria of rats. J Neurosci Res 53, 475–481 (1998).
Dautry, C. et al. Early N-acetylaspartate depletion is a marker of neuronal dysfunction in rats and primates chronically treated with the mitochondrial toxin 3-nitropropionic acid. J. Cereb. Blood. Flow. Metab 20, 789–799 (2000).
Bustillo, J. R. et al. Proton magnetic resonance spectroscopy during initial treatment with antipsychotic medication in schizophrenia. Neuropsychopharmacology 33, 2456–2466 (2008).
Szulc, A. et al. The effect of risperidone on metabolite measures in the frontal lobe, temporal lobe and thalamus in schizophrenic patients. A proton magnetic resonance spectroscopy (1H MRS) study. Pharmacopsychiatry 38, 214–219 (2005).
Szulc, A. et al. The influence of atypical antipsychotics on brain functioning in schizophrenia. A proton magnetic resonance study. Psychiatr. Pol 44, 415–426 (2010).
Deng-Bryant, Y. et al. Ketogenic diet prevents alterations in brain metabolism in young but not adult rats after traumatic brain injury. J. Neurotrauma 28, 1813–1825 (2011).
De Stefano, N. et al. Reversible decreases in N-acetylaspartate after acute brain injury. Magn Reson Med 34, 721–727 (1995).
Miyamoto, S. et al. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol. Psychiatry 10, 79–104 (2005).
Szulc, A. et al. The effect of risperidone on metabolite measures in the frontal lobe, temporal lobe and thalamus in schizophrenic patients. A proton magnetic resonance spectroscopy (1H MRS). Pharmacopsychiatry 38, 214–219 (2005).
Ertugrul, A. et al. The effect of clozapine on regional cerebral blood flow and brain metabolite ratios in schizophrenia: relationship with treatment response. Psychiatry. Res 174, 121–129 (2009).
Braus, D. F. et al. Functioning and neuronal viability of the anterior cingulate neurons following antipsychotic treatment: MR-spectroscopic imaging in chronic schizophrenia. Eur Neuropsychopharmacol 12, 145–152 (2002).
Jarskog, L. F. et al. Effects of davunetide on N-acetylaspartate and choline in dorsolateral prefrontal cortex in patients with schizophrenia. Neuropsychopharmacology 38, 1245–1252 (2013).
Moffett, J. R. et al. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog. Neurobiol. 81, 89–131 (2007).
Nagahama, Y. et al. Cerebral activation during performance of a card sorting test. Brain 119 (Pt 5), 1667–1675 (1996).
Kay, S. R. et al. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr. Bull 13, 261 (1987).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No.81271484 and 81471361 to X.C., Grant No.30900486 and 81371480 to J.T.) and the National Key Basic Research and Development Program (973) (Grant No. 2012CB517904 to X.C.).
Author information
Authors and Affiliations
Contributions
X.Z., J.T., M.H., L.L. and X.C. designed the study. M.H., X.Z., Z.L. and Y.H. collected the samples and participants' characteristics. D.S., H.Z., M.H. and X.Z. carried out the MRS scanning. J.T., Y.L., M.H., X.Z. and J.Z. analyzed and discussed the experimental result. X.Z., J.T., M.H., X.C. and H.C. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zong, X., Hu, M., Li, Z. et al. N-Acetylaspartate Reduction in the Medial Prefrontal Cortex Following 8 weeks of Risperidone Treatment in First-Episode Drug-Naïve Schizophrenia Patients. Sci Rep 5, 9109 (2015). https://doi.org/10.1038/srep09109
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09109
This article is cited by
-
Longitudinal changes in brain metabolites in healthy controls and patients with first episode psychosis: a 7-Tesla MRS study
Molecular Psychiatry (2023)
-
Diverse definitions of the early course of schizophrenia—a targeted literature review
npj Schizophrenia (2018)
-
N-Acetyl-Aspartate in the dorsolateral prefrontal cortex in men with schizophrenia and auditory verbal hallucinations: A 1.5 T Magnetic Resonance Spectroscopy Study
Scientific Reports (2018)
-
Progressive Grey Matter Volume Changes in Patients with Schizophrenia over 6 Weeks of Antipsychotic Treatment and Their Relationship to Clinical Improvement
Neuroscience Bulletin (2018)
-
A Review of the Functional and Anatomical Default Mode Network in Schizophrenia
Neuroscience Bulletin (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
