
Abstract
Therapeutic strategies based on the principles of tissue engineering by self-assembly put forward the notion that functional regeneration can be achieved by utilising the inherent capacity of cells to create highly sophisticated supramolecular assemblies. However, in dilute ex vivo microenvironments, prolonged culture time is required to develop an extracellular matrix-rich implantable device. Herein, we assessed the influence of macromolecular crowding, a biophysical phenomenon that regulates intra- and extra-cellular activities in multicellular organisms, in human corneal fibroblast culture. In the presence of macromolecules, abundant extracellular matrix deposition was evidenced as fast as 48 h in culture, even at low serum concentration. Temperature responsive copolymers allowed the detachment of dense and cohesive supramolecularly assembled living substitutes within 6 days in culture. Morphological, histological, gene and protein analysis assays demonstrated maintenance of tissue-specific function. Macromolecular crowding opens new avenues for a more rational design in engineering of clinically relevant tissue modules in vitro.
Similar content being viewed by others

Introduction
Tissue engineering by self-assembly1,2, cell-sheet tissue engineering3, scaffold-free tissue engineering4 or modular tissue engineering5,6 utilise nature's sophistication to create bottom up supramolecular assemblies. The rationale of either of these approaches lays on the fact that cells are the traditional extracellular matrix (ECM) builders and create tissue modules with precision and stoichiometric competence still unmatched by man-made devices. Such technologies have led to the development of cohesive cell-assembled prototypes, held together by cell-cell and cell-ECM junctions, for skin7,8,9, blood vessel10,11 and cornea12 that have demonstrated reparative/regenerative efficacy, even in clinical setting.
Over 30 years have passed since the development of the first tissue equivalent in vitro7,8,9; it is now evidenced that this scientific breakthrough was not followed by a similar magnitude technological advancement. Indeed, we have now recognised that the major culprit in clinical translation and commercialisation of cell-based therapies is the creation of functional in vitro microenvironments that, by imitating features of the tissue from which the cells were extracted from, will facilitate cell phenotype, function and therapeutic potential maintenance ex vivo13. Thus, bioinspired research efforts recruit pioneered technologies in biophysical cues (e.g. surface topography, substrate elasticity), biochemical beacons (e.g. oxygen tension, ascorbic acid supplementation) and biological signals (e.g. growth factor supplementation, co-culture systems) to maintain permanently differentiated cells phenotype and/or to accurately direct stem cells lineage commitment for the creation of functional tissue facsimiles in vitro14,15,16,17.
Despite the significant advancements that have been achieved to-date, the development of an implantable device remains notoriously difficult and prohibitively expensive, as markedly long ex vivo culture times are required (e.g. 196 days for blood vessel)10 that are associated with loss of native phenotype and cellular senescence18,19. Herein, we hypothesise that macromolecular crowding (MMC), a biophysical phenomenon known to accelerate biological processes by several orders of magnitude20,21,22, will enhance protein turnover and amplify the production of ECM-rich supramolecularly assembled tissue equivalents. Specifically, in vivo, cells inhabit in the dense intertwined network of the ECM, where the proteinase-specific conversion of procollagen to collagen type I is rapid23. In the dilute culture media, the conversion of water soluble procollagen to water insoluble collagen type I is very slow; thus prolonged culture times are required to create an implantable device (Fig. 1). Thus, the addition of inert macromolecules in the culture media, by imitating the dense extracellular space, will enable the accelerated production of ECM-rich living substitutes (Fig. 1). To validate our hypothesis, human corneal fibroblasts (HCFs) were used for very first time as exemplary and the influence of a Ficoll™ cocktail (FC) as a means of MMC on collagen type I, the most abundant ECM protein of corneal stromal, biosynthesis was assessed.

Schematic representation of how MMC enhances ECM deposition in vitro and its application in the development of ECM-rich cell sheet.
Under standard cell culture conditions, the conversion of the water-soluble procollagen to insoluble collagen is very slow as the proteinases are deactivated before they cleave the specific N- and C- propeptides and/or the procollagen is dissolved before its conversion to collagen. After substantially long culture time, the cells will self-crowd the media and collagen is deposited in the cell layer. The addition of inert polydispersed macromolecules (presented as spheres with different diameters) in the culture media results in most effective volume occupancy; in increased relative density of procollagen and proteinases; in cleavage of the propeptides by their respectful proteinases; and finally in accelerated procollagen conversion to collagen and deposition of the former.
Results
Identification of optimal newborn calf serum (NBCS) concentration for maximum ECM deposition
Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE; Fig. S1) and complementary densitometric analysis (Fig. S2) demonstrated that at all NBCS concentrations collagen type I remained in the media in the absence of FC, whilst in the presence of FC, collagen type I was deposited in the cell layer. It was further observed that collagen type I content remained constant or reduced as a function of increased NBCS concentration at given time point in culture. This reduction in collagen type I content was attributed to the enhanced matrix metalloproteinase-2 (MMP-2) activity, as revealed by gelatin zymography (Fig. S3). The highest (p < 0.0001) collagen type I deposition was achieved after culturing HCFs for 6 days in the presence of FC. No significant difference (p > 0.05) in collagen type I deposition was observed among the different NBCS concentrations (0 to 10%) at any time point tested (2, 4 and 6 days). Phase contrast microscopy (Fig. S4) revealed that HCFs maintained their spindle-shaped morphology for all experimental conditions (e.g. presence/absence of FC; 0%–10% NBCS; 2–6 days in culture). Further, the addition of FC did not affect cell metabolic activity (Fig. S5A) and cell viability (Fig. S5B and Fig. S5C) independently of the experimental condition.
Identification of optimal serum origin for maximum ECM deposition
To avoid xenogeneic contaminants, the influence of human serum (HS), as an alternative to NBCS, was assessed at 0.5%, in presence and absence of FC for 2, 4 and 6 days. SDS-PAGE (Fig. S6) and complementary densitometric analysis (Fig. 2A) showed that in the absence of FC collagen type I remained in the media and its content remained constant as a function of time in culture. This indifference in collagen type I content was attributed to the increased MMP-2 content, as revealed by gelatin zymography (Fig. 2B). In the presence of FC, collagen type I was deposited at the cell layer and its content was increased as a function of time in culture. The highest (p < 0.0001) collagen type I deposition was achieved after culturing HCFs for 6 days in the presence of FC. Significant increase in collagen type I (p < 0.001) deposition was observed in the presence of HS, as compared to NBCS at 4 and 6 days. The increase in collagen deposition in the presence of HS was attributed to the lower MMP-2 content of HS (Fig. S7).

MMC accelerates ECM deposition in HCFs culture in the presence of NBCS and HS.
(A) Densitometric analysis of SDS-PAGE confirmed the high collagen type I deposition in the cell layer as early as 2 days in culture. Collagen type I deposition was consistently increased up to 6 days in culture (longer time point assessed). (B) Gelatin zymography detected the 68–72 kDa proMMP-2 and the 62 kDa MMP-2 active form. Both forms of enzyme were higher in NBCS, which explains the enhanced ECM deposition in the presence of HS. (C) ICC analysis further collaborated the enhanced collagen type I deposition in the presence of FC, whilst fibronectin, the template for collagen deposition, was not affected.
Immunocytochemistry (ICC) analysis (Fig. 2C) further corroborated the high collagen type I deposition in the presence of FC and its relative increase as a function of time in culture, independently of the serum origin. No difference in fibronectin deposition was observed. Phase contrast microscopy (Fig. S8) revealed that HCFs maintained their spindle-shaped morphology for all experimental conditions (e.g. presence/absence of FC; 0.5% NBCS/HS; 2–6 days in culture). Further, the presence of FC did not affect cell metabolic activity (Fig. S9A), cell viability (Fig. S9B and Fig. S9C) and DNA content (Fig. S9D), at any given time point, independently of the serum origin.
Production and characterisation of ECM-rich HCFs supramolecular assemblies
Due to the abundant ECM deposition in the presence of FC, commercially available temperature-responsive N-isopropylacrylamide (NIPAAM) coating was not suitable for the detachment of intact ECM-rich HCFs supramolecular assemblies after 6 days in culture, although cell attachment and growth was not prohibited (Fig. S10). A 65% N-isopropylacrylamide/35% N-tert-butylacrylamide (NTBA) copolymer [poly(NIPAAM-co-NTBA)] allowed attachment (Fig. 3A) and detachment (Fig. 3B) of intact ECM-rich HCFs supramolecular assemblies after 6 days in culture in the presence of FC and 0.5% HS. Auxiliary time-lapse microscopy revealed that due to the abundance in deposited ECM under MMC conditions, the detachment rate of the ECM-rich HCFs supramolecular assemblies was slower than their non-MMC counterparts (Fig. 3C and S11).

A poly(NIPAAM-co-NTBA) temperature-responsive copolymer allowed production and detachment of dense and cohesive corneal stromal layers.
(A) The poly(NIPAAM-co-NTBA) temperature-responsive copolymer allowed attachment and spreading of HCFs under MMC and non-MMC conditions. (B) The poly(NIPAAM-co-NTBA) temperature-responsive copolymer allowed intact detachment of the de novo produced ECM-rich HCFs sheets after 6 days in culture. (C) The presence of FC, which resulted in enhanced ECM deposition, delayed the intact detachment of the ECM-rich HCFs sheets; nonetheless, complete detachment achieved within 45 min after temperature reduction.
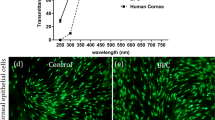
Histological analysis using van Gieson's (Fig. 4A) and Masson's trichrome (Fig. 4B) staining and ICC analysis (Fig. 4C) further corroborated the enhanced collagen type I deposition under MMC conditions (0.5% HS, FC, 6 days in culture). Transmittance analysis (Fig. 4D) revealed no significant difference (p > 0.05) between the groups (non-MMC, 0.5% HS, 6 days in culture; MMC, 0.5% HS, 6 days in culture; PBS; control surface). Further atomic force microscopy analysis (Fig. 4E and S12) revealed the presence of abundant quartered staggered collagen type I fibrils at the intercellular regions of the MMC HCFs (0.5% HS, 6 days in culture).

MMC allowed production of HCFs sheets with physiological, tissue-like characteristics.
(A and B) Histological analysis using van Gieson's and Masson's trichrome staining demonstrate enhanced ECM deposition and tissue-like organization of the HCFs substitutes under MMC conditions. (C) ICC analysis for collagen type I further corroborates the enhanced collagen type I deposition under MMC conditions. (D) No significant difference in transparency between MMC HCFs sheets and PBS, control surface and non-MMC sheets was observed at 0.5% HS and after 6 days in culture. (E) AFM analysis at the intracellular regions (shown are amplitude images) further confirmed the high deposition of collagenous proteins exhibiting characteristic D-periodicity in the presence of FC.
MMC (0.5% HS, FC) did not affect (p > 0.05) gene expression analysis (Fig. 5) for collagens (I, III, IV, V, VI); fibronectin; phenotypic markers (CD34, CHST6); and unintended differentiation marker (α SMA) for all time points assessed (2, 4 and 6 days). Proteomics analysis revealed that under MMC conditions (0.5% HS, FC, 6 days), the total protein content in the cell layer was significantly increased (Table S2). Subsequent validation using ICC (Fig. 6) and complementary fluorescence intensity (Fig. S13) further confirmed the high deposition of collagenous proteins (I, III, IV, V, VI) and fibronectin under MMC conditions (0.5% HS, FC, 6 days in culture), whilst no difference was observed for unintended differentiation markers (α SMA) and phenotypic markers (CD34, keratocan; Fig. S14) for all time points (2, 4 and 6 days in culture).

RT-qPCR analysis demonstrated that MMC did not affect gene expression profile of HCFs at respective time points.
The expression of collagens (I, III, IV, V, VI); glycoproteins (fibronectin); phenotypic markers (CD34, CHST6); and unintended differentiation markers (α SMA) for all time points assessed (2, 4 and 6 days) remained unaffected under the conditions tested (0.5% HS, with or without FC).

MMC enhanced ECM deposition in the cell layer of HCFs.
Validation of proteomics analysis with ICC further confirmed the high deposition of collagenous proteins (I, III, IV, V, VI) and glycoproteins (fibronectin) in the presence of FC, whilst no difference was observed for unintended differentiation markers (α SMA) at all time points (2, 4 and 6 days). A high level of structural alignment is also evidenced.
Low-density versus high-density culture
At day 6, MMC at low-density (25,000 cells/cm2) culture induced higher ECM deposition than non-crowded high-density (50,000 cells/cm2) culture (Fig. 7). The highest ECM deposition was detected in the high-density culture in the presence of MMC (Fig. 7).

MMC is more effective in producing ECM-rich living substitutes than high-density non-crowded cultures.
At day 6, MMC at low-density (LD; 25,000 cells/cm2) culture induced higher ECM deposition than non-crowded high-density (HD; 50,000 cells/cm2) culture and the highest ECM deposition was detected in the high-density culture in the presence of MMC, as evidenced by SDS-PAGE (A) and complementary densitometric (B) analysis, immunocytochemistry analysis (C, D, E) and staining assays (F, G).
Discussion
During development, cellular secretome creates a tissue-specific microenvironment that governs the various functions of the tissue. Cellular secretome also provides necessary signals for tissue repair and regeneration during or following injury. Therapeutic strategies based on the principles of tissue engineering by self-assembly aim to capture this specific reparative potential of cellular secretome. However, for such approaches to succeed, it is essential to create a functional ex vivo microenvironment that by emulating the tissue from which the cells were extracted from or are to be implanted in will maintain cell phenotype, function and therapeutic potential. Unfortunately, the still primitive culture conditions not only are associated with phenotypic drift, but also require a prolonged period of time for the development of an implantable device. Herein, we hypothesised that MMC, by imitating the dense extracellular space, will accelerate the deposition of tissue-specific ECM, whilst preserving cell phenotype. Given that tissue engineering by self-assembly strategies for corneal stromal require 4 to 8 weeks for the production of supramolecularly assembled monolayers24,25,26 and the readiness of corneal fibroblasts to trans-differentiate towards unintended myofibroblast lineage27,28,29,30,31,32, we used this cell type as model.
In the absence of MMC, collagen type I remained in the media, whilst in the presence of FC ample ECM deposition was evidenced in the cell layer as early as 48 h in culture, which persisted up to 6 days in culture (longer time point tested). This is in accordance to previous observations, where FC accelerated ECM deposition in human lung fibroblast culture33 and human bone marrow stem cell culture34. The efficacy of FC to induce accelerated ECM deposition lays on the fact that effectively excludes volume35,36, facilitating that way cleavage of C- and distant N-propeptides by their respective proteinases and therefore accelerated conversion of procollagen to collagen type I.
Tissue engineering by self-assembly supramolecular aggregates are customarily produced using copious amounts of animal sera (up to 30%). Our data indicate that excessive sera supplementation is counter-effective, due to the high amount of MMP-2 present that degrades deposited ECM. Given though that sera contain survival signals, mitogens and growth factors, all necessary for cell expansion, a balance needs to be achieved that will allow both cell survival and ECM deposition. Herein, we demonstrate that low serum concentration appears to achieve optimal balance between matrix deposition and cell growth, which is in accordance to previous results, where high serum supplementation resulted in phenotypic drift of corneal stromal cells37,38,39, tenocytes40 and retinal progenitor cells41, whilst low serum or serum-free media resulted in phenotype maintenance of dental-derived stem cells42, embryonic stem cells43 and mesenchymal stem cells44,45. Further, given the potential of interspecies transmission of disease and severe immune reactions associated with animal sera, our data further corroborate previous studies supporting the use of HS for clinically relevant cell therapies46,47,48,49,50.
The ample deposition of ECM in the presence of FC prohibited harvesting of intact supramolecularly assembled ECM-rich HCFs substitutes from the brush-like commercially available NIPAAM dishes. The efficiency of poly(NIPAAM-co-NTBA) copolymer to produce intact ECM-rich cell sheets is attributed to the decreased number of N – H residues, due to the additional steric hindrance induced by the addition of the NTBA group, which decreased hydrogen bonding and consequently decreased protein adsorption51, making cell detachment easier. Although the detachment rate of the MMC samples was slower than the non-MMC counterparts, a fully characterised poly(NIPAAM-co-NTBA) copolymer52,53,54 allowed harvesting of dense and cohesive tissue-like modules as a continuous sheet within 45 min by a simple switch of temperature from 37°C to 10°C. Subsequent morphological analysis revealed that these dense and cohesive supramolecularly assembled living substitutes produced within 6 days in culture in the presence of FC had intact cell-cell and cell-ECM junctions and achieved similar level of biomimicry as scaffold and scaffold-free substitutes that were produced after 355, 424, 956 or even 1157 weeks in culture. Further, although traditional self-assembled living substitutes require prolonged culture time to form and mature, in the presence of macromolecular crowding, such processes are enhanced, as evidenced by the presence of β-bands on the gels.
Further gene and protein analysis assays demonstrated that FC did not affect expression levels of collagenous proteins, glycoproteins, phenotypic markers and unintended differentiation markers. It is worth pointing out that both MMC and non-MMC counterparts were negative for CD34 and keratocan at protein level and very low expression at gene level. It has been reported that the expression of these markers rapidly declines in low-density cultures58 after exposure to serum37,38,39,59 and as a function of time in culture56,60,61.
Here, we demonstrate that modulation of the in vitro microenvironment of HCFs with FC results in ECM-rich supramolecular assemblies within days rather than weeks or months in culture, without compromising fundamental cellular functions. This technology not only requires a lower cell number than multilayer cell sheets or high-density cultures, which is often not available, but also bypasses altogether such approaches that due to poor nutrient, oxygen and waste transport result in cell necrosis in the central cell-layers62. It further evades complex and expensive culture media that only temporarily will maintain phenotype, without actually increasing ECM deposition63,64,65,66,67.
Methods
Materials
Tissue culture consumables were purchased from Sarstedt (Ireland) and Nunc (Denmark). All other materials, including cell culture media, reagents and macromolecular crowders (Ficoll™ 70, Ficoll™ 400) were purchased from Sigma Aldrich (Ireland), unless otherwise stated. Live/Dead® viability/cytotoxicity kit and alamarBlue® cell metabolic activity kit were purchased from BioSource International, Invitrogen (Ireland).
Corneal fibroblast culture
Primary HCFs (Innoprot, Spain) were cultured as per supplier protocol. Briefly 5,000 cells/cm2 were seeded on poly (L-lysine) coated tissue culture flasks maintained at 37°C with 5% CO2/95% air in a humidified incubator. Cells used in all experiments were between 3 and 5 passage.
Macromolecular crowding treatment
HCFs were seeded in 24-well plates at 25,000 cells/cm2 density. When we compared low-density and high-density cultures, 25,000 cells/cm2 and 50,000 cells/cm2 were used, respectively. The next day, the media was replaced with fresh containing FC (Ficoll™ 70 + Ficoll™ 400: 37.5 mg/ml + 25 mg/ml) in the presence of 0.0 to 10.0% NBCS or HS. 100 μM L ascorbic acid phosphate supplement was added in the media during the crowding experiment to enhance ECM synthesis. The influence of MMC was assessed at 2, 4 and 6 days in culture.
Collagen deposition analysis
Media and cell layers were digested with porcine gastric mucosa pepsin (Sigma Aldrich, Ireland) for 2 h at 37°C with continuous shaking and subsequent neutralisation with 1 N NaOH. The samples for SDS-PAGE were prepared using appropriate dilution with distilled water and 5 × sample buffer. Finally, 15 μl per sample solution per well was loaded on the gel (5% running gel/3% stacking gel) after 5 min heating at 95°C. Electrophoresis was performed in a Mini-PROTEAN Tetra Electrophoresis System (Bio-Rad, Ireland) by applying potential difference of 50 mV for the initial 30 min and then 120 mV for the remaining time (approximately 1 h). The gels were washed gently with ultra pure water and stained using silver stain kit (SilverQuest™, Invitrogen, Ireland) according to the manufacturer's protocol. Images of the gels were taken after brief washing with water. In order to quantify the cell-produced collagen type I deposition, the relative densities (GeneTools software, Syngene, Ireland) of collagen α1(I) and α2(I) chains were evaluated and compared to the α1(I) and α2(I) chain bands densities of standard collagen type I (Symatese Biomateriaux, France).
Matrix metalloproteinase analysis
The presence of MMPs was evaluated using gelatin zymography. Briefly, at the end of each cell culture time point, media were collected and mixed with non-reducing SDS sample buffer (125 mM Tris-HCl, pH 6.8; 20% glycerol; 2% SDS; 0.002% bromophenol blue) and fractionated by SDS-PAGE using 10% gels containing 0.1% gelatin. After electrophoresis, the gels were washed with two incubations in 2.5% Triton X-100 for 30 min. The gels were incubated for 18 h at 37°C in a reaction buffer containing 50 mM Tris, pH 7.4; 5 mM CaCl2; 1 μM ZnCl2 to promote recovery of protease activity were and then stained with 0.5% Coomassie G250 brilliant blue for 30 min. Images of the gels were taken after de-staining with 30% ethanol/10% acetic acid. These gelatin zymography gels bands were compared for relative expression of MMP-2. Uncultured media with various percentages of NBCS and HS were also used as controls in respective experiments.
Cell morphology analysis
Cell morphology was assessed using phase contrast microscopy (Olympus IX81 inverted microscope, Japan).
Cell metabolic activity and viability assessment
The influence of MMC on cell metabolic activity and viability was assessed using alamarBlue® and Live/Dead® assays, as per manufacturer's guidelines. Briefly, cell culture media containing 10% alamarBlue® reagent was added to various samples after removing the culture medium and brief washing with PBS and then incubated for 4 h to allow for the reduction of resazurin dye by active cells in cellular metabolism assay. Following incubation, 100 μl of medium samples were transferred into a black 96-well plate. Fluorescence of the media was determined using a micro-plate reader (Varioskan Flash, Thermo Scientific, UK) at excitation and emission 570 nm/600 nm wavelength. The level of metabolic activity was calculated using % reduction of dye, according to the supplier's protocol and compared with the respective control samples. The cell viability was determined using Live/Dead® viability kit (Invitrogen, Ireland). The cells were incubated with calcium AM and ethidium homodimer solution (2 μM calcein-AM and 4 μM EthD-1) in PBS according to manufacturer's staining protocol for at least 30 min. The cell layers were washed in fresh PBS to remove excess dye. Following that, fluorescence images were taken using an Olympus IX81 inverted fluorescence microscope.
DNA quantification
DNA quantification was carried out using Quant-iT™ PicoGreen® dSDNA assay kit (Invitrogen, Ireland) according to the manufacturer's protocol. Briefly, DNA was extracted using three freeze-thaw cycles after adding 200 μl of nucleic acid free water per well (24 well plate). The cell suspension was subsequently transferred to cold eppendorf tubes and was centrifuged for 5 minutes at 12000 rpm. 25 μl were then transferred into 96-well plate containing 75 μl of 1 × TE buffer. A standard curve was generated using 0, 7.8, 15.6, 31.2, 62.5, 125, 250 and 500 μg/mL DNA concentrations. 100 μl of a 1:200 dilution of Quant-iT™ PicoGreen® reagent was added to each sample and the plate was read using a micro-plate reader (Varioskan Flash, Thermo Scientific, Ireland) with an excitation wavelength of 480 nm and an emission wavelength of 525 nm.
Immunocytochemistry analysis
Cells were seeded in 4-well-chamber Lab-Tek™ II slide and MMC was carried out as described above. At the end of each culture time point, cells were fixed with 3% formaldehyde in PBS (pH 7.4). After fixation, the specimens were washed three times in PBS. The fixed cell layers were incubated with 3% BSA for 30 min to stop the non-specific binding of proteins. Cell layers were incubated with primary antibodies diluted in PBS for 90 min at room temperature. Rabbit collagen type I, III, IV, V and VI (Abcam, UK; 1:200) and mouse anti-fibronectin antibody (Sigma Aldrich, Ireland; F7387 1:200) were used to detect the expression of various collagens and fibronectin respectively. Mouse anti-actin, α smooth muscle antibody (1:400 dilution), mouse CD34 antibody (1:50 dilution) and mouse keratocan antibody (1:50 dilution) were used to detect α SMA, CD34 and keratocan expression in HCFs culture. Methanol fixing (cold methanol at −20°C for 5 min) was also used after fixation to detect CD34. After primary incubation, the samples were incubated with secondary antibody for 30 min at room temperature. The secondary antibodies used were Alexa Fluor® 488, chicken anti rabbit or donkey anti mouse respectively for rabbit and mouse antibodies at the 1:400 dilutions (Invitrogen, Ireland). Antibody incubation was followed by three washes in PBS. For nuclear staining, DAPI (4′,6-diamidino-2-phenylindole) was used at 1:4000 dilution (Invitrogen, Ireland). Finally, the cover slips were mounted on glass slides with VectaShield (Vector Laboratories, UK) for direct observation. The images were taken using an Olympus IX81 inverted fluorescence microscope using 10 × objective. The intensity of the fluorescence was evaluated using the Scope-Pro Plus software.
Cell sheet production
Poly(NIPAAM-co-NTBA) was dissolved in anhydrous ethanol at 40 μg/ml and subjected to continuous shaking overnight. This acrylamide solution was mixed with poly(L-lysine) (100 μg/ml) in 1:1 V/V ratio and again allowed to shake overnight to mix properly. 100 μl of the mixed solution was deposited onto each of the petri dishes, which were left in ethanol soaked desiccator overnight. The dishes were further dried in 600 mBar vacuum oven at 40°C for at least 4 h. Cell culture was carried out as described above after mild UV sterilisation of the petri dishes for 2 h. Following culture, a temperature-controlled plate was used to induce detachment and subsequent cell-sheet harvesting.
Histological analysis using van Gieson staining
After 6 days in culture, samples were fixed with 3% paraformaldehyde for at least 30 min. The samples were then incubated with Weigert working solution for 10 min after 3 brief PBS washings. Finally, the samples were incubated in van Gieson's solution for 2–3 min, after brief PBS washing. The samples were dehydrated using 95% alcohol, absolute alcohol and xylene. Images were taken using the Olympus microscope (BX 51) at 100×.
Histological analysis using Masson's Trichrome staining
Samples were fixed in Bouin's solution for 1 h at 56°C after fixation. The samples were then incubated in Weigert's iron haematoxylin staining for 10 min, followed by rinsing with running tap water. The samples were then washed with distilled water, after 10 min in warm running tap water. The samples were incubated in Biebrich scarlet-acid fuchsin solution for 10 to 15 min and washed again with distilled water. Phosphomolybdic-phosphotungstic acid solution was used for further 15 min and the samples were then transferred directly to aniline blue solution and stained for 10 min, rinsed briefly in distilled water and then in 1% acetic acid solution for 5 min. The samples were dehydrated quickly through 95% ethyl alcohol, absolute ethyl alcohol (these step wipes off Biebrich scarlet-acid fuchsin staining) and cleared in xylene after brief washing in distilled water. Images were then taken using the Olympus inverted microscope (BX 51) at × 100.
Atomic force microscopy analysis
HCFs were seeded in 4-well-chamber Lab-Tek™ II slides and cell culture took place as described above. After six days in culture, cell layers were washed with HBSS and fixed at room temperature for 15 min. The cell layers were then washed three times with PBS and serially dehydrated with 30%, 50%, 70%, 90% and 100% ethanol. Atomic force microscopy (MFP-3D, Asylum Research, USA) analysis was then performed using rectangular Si cantilevers (SSS-NCH, Nanosensors, Switzerland), each having a nominal resonance frequency of 330 kHz and a spring constant of 42 N/m. Images were recorded using amplitude modulation mode in an ambient environment after drying the samples with nitrogen.
Light transmission
Light transmission was assessed using a previously described method26. Briefly, the media was aspirated and 100 μl PBS was added in each sample well after PBS washing. PBS alone was used as control blank. For zero absorbance and 100% absorbance, the wells without PBS or cells and black dye were taken as control; this black dye showed approximately 100% absorbance (0% transmittance), while the absorbance value in air was taken as 100% transmittance. The optical density of the samples was measured using a spectrophotometer (Varioskan Flash, Thermo Scientific, Ireland) at 380–780 nm wavelength with a resolution of 5 nm. The % transmission was calculated as: % Transmittance = 100 (10ˆ-A), where A is absorbance measured.
Gene expression (RT-qPCR) analysis
Total RNA was extracted using a modified Trizol isolation method at the given time points. Briefly, TriReagent® (Invitrogen, Ireland) was added to the cell layer after aspiration of medium from the culture and brief washing with PBS. The cellular layer was mechanically disrupted using gentle pipetting of tissue culture plate. Phase separation was conducted with chloroform and the total RNA contained in the aqueous phase was purified using RNeasy® mini kit column (QIAGEN, Hilden, Germany), according to the supplier's protocol. Three extractions were carried out for each sample and pooled at the end of the RNeasy protocol. The purity and quantity of total RNA were evaluated using an ultraviolet spectrometer (NanoDrop ND-1000 Spectrophotometer, Thermo Scientific, Ireland). Reverse transcription (RT) was performed using MJ Research PTC-200 DNA Engine system according to the manufacturer's protocol (Promega RT System, UK). The prepared cDNA was monitored using SYBRGreen master mix (QIAGEN, Hilden, Germany) by real-time PCR using StepOnePlus™ Real-Time PCR System (Applied Bioscience, Switzerland). Gene transcription was normalised to the transcription of housekeeping human 18S gene. The 2−ΔΔCt method was used to calculate relative gene expression for each target gene at respective time point. Primers used for various collagens, fibronectin, CD34, CHST6 and α SMA are given in Table S1.
Proteomics analysis
Total protein extraction from the cell layers was carried out using Qproteome™ mammalian protein preparation kit (Qiagen, UK). Briefly, cell layers were washed twice with PBS and scraped gently, using a cell scraper, in the presence of ice cold PBS and transferred to pre-chilled 1.5 ml tubes. This solution was centrifuged at 450 g for five min at 4°C. The cell layer was lysed gently with cell lysis buffer containing Benzonase® nuclease and protease inhibitor after discarding the supernatant. This suspension was centrifuged again at 14,000 g at 4°C for 10 min, after agitation in a rotary shaker for 5 min. After centrifugation, the supernatant was transferred into pre-chilled 0.5 ml tubes (Protein LoBind Tubes, UK) and freeze-dried. High-throughput proteomic profiling was performed using the 4-plex iTRAQ labelling kit, equipped with the 4700 MALDI TOF/TOF system (Applied Biosystems, USA). Briefly, the protein pellet from each sample was re-suspended in a protein dissolution buffer (10 mM triethylammonium bicarbonate, pH 8.5) and each protein sample was assigned to one of four isobaric tags as per the manufacturer's instructions. The tagged samples were pooled together and subjected to mass spectrometry analysis. Samples were fractionated using strong cation exchange and separated by reverse phase chromatography using the UltimatePlus NanoLC system (Dionex, USA). After reverse phase chromatography, the eluted protein fractions were spotted onto a MALDI plate (ABI 4800 OptiTOF, Applied Biosystems, USA). Using a Probot printing robot (Dionex, USA), the spotted plate was mixed with α-Cyano-4-hydroxycinnamic acid ionization matrix (Sigma Aldrich, Ireland) at a ratio of 1:2. Mass spectrometry analysis of the spotted plate was carried out an ABI 4800Plus MALDI-TOF/TOF tandem MS system (Applied Biosystems, USA). The time-of-flight of the protein was proportional to the molecular weight. The final read-out graph was a multi-peak spectrum with different peak intensities, which correspond to the relative protein amount. The peak location corresponds to the precise protein or peptide molecular weight. Data analysis was performed using Protein Pilot 2.0 software (Applied Biosystems, USA) on SWISS-PROT, TrEMBL (www.ebi.ac.uk/swissprot) and NCBI (www.ncbi.nlm.nih.gov/) non-redundant protein databases. The unused score was kept at more than 1.3 to get the confidence interval more than 90; relative peptide value at 95% was compared among samples. Selected ECM proteins detected in proteomics results were validated using ICC and fluorescent intensity, using Olympus IX81 inverted microscope.
Statistical analysis
All results presented are mean ± SD. Statistical analysis (MINITAB™ version 16, Minitab, Inc., USA) was performed using two-sample t-test for pair wise comparisons or one away analysis of variance (ANOVA) for multiple comparisons after confirming: (a) the normal distribution from which each of the samples (Anderson-Darling normality test); and (b) the variances of the population of the samples were equal to one another (Bartlett's and Levene's tests for homogenicity of variance). Non-parametric statistics were used if above assumptions were violated and consequently Kruskal-Wallis test for multiple comparisons or Mann-Whitney test for 2-samples used. Each experiment was performed in biological triplicates in minimum three samples. Differences between selective experimental groups were considered statistically significant at p value < 0.05.
References
Guillame-Gentil, O. et al. Engineering the extracellular environment: Strategies for building 2D and 3D cellular structures. Adv Mater 22, 5443–5462 (2010).
Peck, M., Dusserre, N., McAllister, T. N. & L'Heureux, N. Tissue engineering by self-assembly. Mater Today 14, 218–224 (2011).
Matsuda, N., Shimizu, T., Yamato, M. & Okano, T. Tissue engineering based on cell sheet technology. Adv Mater 19, 3089–3099 (2007).
Kelm, J. M. & Fussenegger, M. Scaffold-free cell delivery for use in regenerative medicine. Adv Drug Deliv Rev. 62, 753–764 (2010).
Dankers, P., Harmsen, M., Brouwer, L., Luyn, M. V. & Meijer, E. A modular and supramolecular approach to bioactive scaffolds for tissue engineering. Nat Mater 4, 568–574 (2005).
Nichol, J. W. & Khademhosseini, A. Modular tissue engineering: Engineering biological tissues from the bottom up. Soft Matter 5, 1312–1319 (2009).
Green, H., Kehinde, O. & Thomas, J. Growth of cultured human epidermal cells into multiple epithelia suitable for grafting. Proc Natl Acad Sci U S A 76, 5665–5668 (1979).
Gallico, G., O'Connor, N., Compton, C., Kehinde, O. & Green, H. Permanent coverage of large burn wounds with autologous cultured human epithelium. N Engl J Med. 311, 448–451 (1984).
Phillips, T., Kehinde, O., Green, H. & Gilchrest, B. Treatment of skin ulcers with cultured epidermal allografts. J Am Acad Dermatol. 21, 191–199 (1989).
L'Heureux, N. et al. Human tissue-engineered blood vessels for adult arterial revascularization. Nat Med. 12, 361–365 (2006).
L'Heureux, N., McAllister, T. N. & de la Fuente, L. M. Tissue-engineered blood vessel for adult arterial revascularization. N Engl J Med. 357, 1451–1453 (2007).
Nishida, K. et al. Corneal reconstruction with tissue-engineered cell sheets composed of autologous oral mucosal epithelium. N Engl J Med. 351, 1187–1196 (2004).
Cigognini, D. et al. Engineering in vitro microenvironments for cell based therapies and drug discovery. Drug Discov Today 18, 1099–1108 (2013).
Bates, C. J., Bailey, A. J., Prynne, C. J. & Levene, C. I. The effect of ascorbic acid on the synthesis of collagen precursor secreted by 3T6 mouse fibroblasts in culture. Biochim Biophys Acta. 278, 372–390 (1972).
Engler, A. J., Sen, S., Sweeney, H. L. & Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006).
Ivanovic, Z. Hypoxia or in situ normoxia: The stem cell paradigm. J Cell Physiol. 219, 271–275 (2009).
Edalat, F., Bae, H., Manoucheri, S., Cha, J. & Khademhosseini, A. Engineering approaches toward deconstructing and controlling the stem cell environment. Ann Biomed Eng. 40, 1301–1315 (2012).
Campisi, J. & Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat Rev Mol Cell Biol 8, 729–740 (2007).
Beltrami, A., Cesselli, D. & Beltrami, C. Stem cell senescence and regenerative paradigms. Clin Pharmacol Ther. 91, 21–29 (2012).
Zimmerman, S. B. & Harrison, B. Macromolecular crowding increases binding of DNA polymerase to DNA: An adaptive effect. Proc Natl Acad Sci U S A 84, 1871–1875 (1987).
Minton, A. P., Colclasure, G. C. & Parker, J. C. Model for the role of macromolecular crowding in regulation of cellular volume. Proc Natl Acad Sci U S A 89, 10504–10506 (1992).
Zhou, Z. et al. Crowded cell-like environment accelerates the nucleation step of amyloidogenic protein misfolding. J Biol Chem. 284, 30148–30158 (2009).
Canty, E. G. & Kadler, K. E. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. 118, 1341–1353 (2005).
Guo, X. et al. Morphologic characterization of organized extracellular matrix deposition by ascorbic acid–stimulated human corneal fibroblasts. Invest Ophthalmol Vis Sci. 48, 4050–4060 (2007).
Proulx, S. et al. Reconstruction of a human cornea by the self-assembly approach of tissue engineering using the three native cell types. Mol Vis. 16, 2192–2201 (2010).
Grobe, G. M. & Reichl, S. Characterization of vitamin C-induced cell sheets formed from primary and immortalized human corneal stromal cells for tissue engineering applications. Cells Tiss Org. 197, 283–297 (2013).
Jester, J. V., Petroll, W. M., Barry, P. A. & Cavanagh, H. D. Expression of alpha-smooth muscle (alpha-SM) actin during corneal stromal wound healing. Invest Ophthalmol Vis Sci. 36, 809–819 (1995).
Fini, M. E. Keratocyte and fibroblast phenotypes in the repairing cornea. Prog Retin Eye Res. 18, 529–551 (1999).
Espana, E., Kawakita, T., Liu, C. & Tseng, S. CD-34 expression by cultured human keratocytes is downregulated during myofibroblast differentiation induced by TGF-beta1. Invest Ophthalmol Vis Sci. 45, 2985–2991 (2004).
Pei, Y., Sherry, D. M. & McDermott, A. M. Thy-1 distinguishes human corneal fibroblasts and myofibroblasts from keratocytes. Exp Eye Res. 79, 705–712 (2004).
Yoshida, S., Shimmura, S., Shimazaki, J., Shinozaki, N. & Tsubota, K. Serum-free spheroid culture of mouse corneal keratocytes. Invest Ophthalmol Vis Sci. 46, 1653–1658 (2005).
West-Mays, J. A. & Dwivedi, D. J. The keratocyte: Corneal stromal cell with variable repair phenotypes. Int J Biochem Cell Biol. 38, 1625–1631 (2006).
Lareu, R. R. et al. Collagen matrix deposition is dramatically enhanced in vitro when crowded with charged macromolecules: The biological relevance of the excluded volume effect. FEBS Lett. 581, 2709–2714 (2007).
Zeiger, A. S., Loe, F. C., Li, R., Raghunath, M. & Van Vliet, K. J. Macromolecular crowding directs extracellular matrix organization and mesenchymal stem cell behavior. Plos One 7, e37904 (2012).
Satyam, A. et al. Macromolecular crowding meets tissue engineering by self-assembly: A paradigm shift in regenerative medicine. Adv Mater 26, 3024–3034 (2014).
Zeugolis, D. & Satyam, A. inventors. National Universty of Ireland, Galway, assignee. Engineered living tissue substitute. European patent EP 2 532 736. 2012 December 12.
Funderburgh, J. L., Mann, M. M. & Funderburgh, M. L. Keratocyte phenotype mediates proteoglycan structure: A role for fibroblasts in corneal fibrosis. J Biol Chem. 278, 45629–45637 (2003).
Guerriero, E. et al. Loss of alpha3(IV) collagen expression associated with corneal keratocyte activation. Invest Ophthalmol Vis Sci. 48, 627–635 (2007).
Kawakita, T. et al. Keratocan expression of murine keratocytes is maintained on amniotic membrane by down-regulating transforming growth factor-beta signaling. J Biol Chem. 280, 27085–27092 (2005).
Imada, K. et al. Serum-dependent osteoblastic changes in cultured tenocytes isolated from rat Achilles tendon. J Tokyo Med Uni. 71, 143–150 (2013).
Hu, Y. et al. An in vitro comparison study: The effects of fetal bovine serum concentration on retinal progenitor cell multipotentiality. Neurosc Lett. 534, 90–95 (2013).
Tarle, S. A., Shi, S. & Kaigler, D. Development of a serum-free system to expand dental-derived stem cells: PDLSCs and SHEDs. J Cellul Physiol. 226, 66–73 (2011).
Hannoun, Z. et al. The comparison between conditioned media and serum-free media in human embryonic stem cell culture and differentiation. Cellular Reprogr. 12, 133–140 (2010).
Meuleman, N. et al. Human marrow mesenchymal stem cell culture: Serum-free medium allows better expansion than classical α-MEM medium. Eur J Haemat. 76, 309–316 (2006).
Kuznetsov, S. A., Mankani, M. H. & Robey, P. G. Effect of serum on human bone marrow stromal cells: Ex vivo expansion and in vivo bone formation. Transplant 70, 1780–1787 (2000).
Ang, L., Tan, D., Seah, C. & Beuerman, R. The use of human serum in supporting the in vitro and in vivo proliferation of human conjunctival epithelial cells. Br J Ophthalmol. 89, 748–752 (2005).
Mannello, F. & Tonti, G. Concise review: No breakthroughs for human mesenchymal and embryonic stem cell culture: Conditioned medium, feeder layer, or feeder-free; medium with fetal calf serum, human serum, or enriched plasma; serum-free, serum replacement nonconditioned medium, or ad hoc formula? All that glitters is not gold!. Stem Cells 25, 1603–1609 (2007).
Tateishi, K. et al. Comparison of human serum with fetal bovine serum for expansion and differentiation of human synovial MSC: Potential feasibility for clinical applications. Cell Transplant 17, 549–557 (2008).
Aldahmash, A. et al. Human serum is as efficient as fetal bovine serum in supporting proliferation and differentiation of human multipotent stromal (mesenchymal) stem cells in vitro and in vivo. Stem Cell Rev and Rep. 7, 860–868 (2011).
Dreher, L. et al. Cultivation in human serum reduces adipose tissue-derived mesenchymal stromal cell adhesion to laminin and endothelium and reduces capillary entrapment. Stem Cells Dev. 22, 791–803 (2013).
Lynch, I. et al. Correlation of the adhesive properties of cells to N-isopropylacrylamide/N-tert-butylacrylamide copolymer surfaces with changes in surface structure using contact angle measurements, molecular simulations and Raman spectroscopy. Chem Mater 17, 3889–3898 (2005).
Allen, L. T. et al. Interaction of soft condensed materials with living cells: Phenotype/transcriptome correlations for the hydrophobic effect. Proc Natl Acad Sci U S A 100, 6331–6336 (2003).
Gilcreest, V. et al. Thermoresponsive poly(N-isopropylacrylamide) copolymers: Contact angles and surface energies of polymer films. Langmuir 20, 10138–10145 (2004).
Moran, M. T., Carroll, W. M., Selezneva, I., Gorelov, A. & Rochev, Y. Cell growth and detachment from protein-coated PNIPAAm-based copolymers. J Biomed Mater Res A. 81, 870–876 (2007).
Du, Y. et al. Secretion and organization of a cornea-like tissue in vitro by stem cells from human corneal stroma. Invest Ophthalmol Vis Sci. 48, 5038–5045 (2007).
Wu, W. J. et al. Bioengineering organized, multilamellar human corneal stromal tissue by growth factor supplementation on highly aligned synthetic substrates. Tissue Eng. 19, 2063–2075 (2013).
Ren, R. et al. Human primary corneal fibroblasts synthesize and deposit proteoglycans in long-term 3-D cultures. Developm Dyn. 237, 2705–2715 (2008).
Musselmann, K., Alexandrou, B., Kane, B. & Hassell, J. R. Maintenance of the keratocyte phenotype during cell proliferation stimulated by insulin. J Biol Chem. 280, 32634–32639 (2005).
Kawakita, T. et al. Preservation and expansion of the primate keratocyte phenotype by downregulating TGF-beta signaling in a low-calcium, serum-free medium. Invest Ophthalmol Vis Sci. 47, 1918–1927 (2006).
Builles, N. et al. Development of an optimised culture medium for keratocytes in monolayer. Biomed Mater Eng. 16, S95–S104 (2006).
Perrella, G. et al. Expression of haematopoietic stem cell markers, CD133 and CD34 on human corneal keratocytes. Br J Ophthalmol. 91, 94–99 (2007).
Shimizu, T. et al. Polysurgery of cell sheet grafts overcomes diffusion limits to produce thick, vascularized myocardial tissues. FASEB J. 20, 708–710 (2006).
Pancholi, S., Tullo, A., Khaliq, A., Foreman, D. & Boulton, M. The effects of growth factors and conditioned media on the proliferation of human corneal epithelial cells and keratocytes. Graefes Arch Clin Exp Ophthalmol. 236, 1–8 (1998).
Jester, J. V. & Ho-Chang, J. Modulation of cultured corneal keratocyte phenotype by growth factors/cytokines control in vitro contractility and extracellular matrix contraction. Exp Eye Res. 77, 581–592 (2003).
Etheredge, L. et al. Enhanced cell accumulation and collagen processing by keratocytes cultured under agarose and in media containing IGF-I, TGF-β or PDGF. Matrix Biol. 29, 519–524 (2010).
Kim, A., Lakshman, N., Karamichos, D. & Petroll, W. M. Growth factor regulation of corneal keratocyte differentiation and migration in compressed collagen matrices. Invest Ophthalmol Vis Sci. 51, 864–875 (2010).
Lakshman, N. & Petroll, W. Growth factor regulation of corneal keratocyte mechanical phenotypes in 3-D collagen matrices. Invest Ophthalmol Vis Sci. 53, 1077–1086 (2012).
Acknowledgements
We thank Dr Oliver Carroll for technical support and Mr Maciej Doczyk (http://doczykdesign.com) for the preparation of Fig. 1. This work is supported by Science Foundation Ireland, Research Frontiers Programme, Project Number: SFI-09-RFP-ENM2483 to D.Z.; Science Foundation Ireland, E.T.S. Walton Visitor Awards Programme, Project Number: 08/W.1/B2568 to M.R., A.P. and D.Z.; Health Research Board, Project Number: HRA_POR/2011/84 to D.Z.; and College of Engineering & Informatics, National University of Ireland, Galway, Postgraduate College Fellowship Scheme to P.K. and D.Z.
Author information
Authors and Affiliations
Contributions
P.K. and D.Z. designed research; P.K., A.S., X.F. and E.C. performed experimental work; P.K., A.S., X.F., A.G. and Y.R. developed temperature responsive polymers and cell sheets; P.K., A.S., S.D. and D.Z. designed, performed, analysed and validated the proteomics study; P.K., A.S. and B.J.R. designed, performed and analysed AFM study; P.K., A.S. and D.Z. analysed data; A.P., M.R., L.J. and D.Z. discussed results; P.K. and D.Z. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kumar, P., Satyam, A., Fan, X. et al. Macromolecularly crowded in vitro microenvironments accelerate the production of extracellular matrix-rich supramolecular assemblies. Sci Rep 5, 8729 (2015). https://doi.org/10.1038/srep08729
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08729
This article is cited by
-
Fibroblasts are not just fibroblasts: clear differences between dermal and pulmonary fibroblasts’ response to fibrotic growth factors
Scientific Reports (2023)
-
Macromolecular crowding regulates matrix composition and gene expression in human gingival fibroblast cultures
Scientific Reports (2023)
-
Macromolecular crowding facilitates rapid fabrication of intact, robust cell sheets
Biotechnology Letters (2023)
-
Adipose-derived stromal cells increase the formation of collagens through paracrine and juxtacrine mechanisms in a fibroblast co-culture model utilizing macromolecular crowding
Stem Cell Research & Therapy (2022)
-
Identification of lamprey variable lymphocyte receptors that target the brain vasculature
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
