
Abstract
Tissue-derived decellularized biomaterials are ideal for tissue engineering applications as they mimic the biochemical composition of the native tissue. These materials can be used as hydrogels for cell encapsulation and delivery. The decellularization process can alter the composition of the extracellular matrix (ECM) and thus influence the hydrogels characteristics. The aim of this study was to examine the impact of decellularization protocols in ECM-derived hydrogels obtained from porcine corneas. Porcine corneas were isolated and decellularized with SDS, Triton X-100 or by freeze-thaw cycles. All decellularization methods decreased DNA significantly when measured by PicoGreen and visually assessed by the absence of cell nuclei. Collagen and other ECM components were highly retained, as quantified by hydroxyproline content and sGAG, by histological analysis and by SDS-PAGE. Hydrogels obtained by freeze-thaw decellularization were the most transparent. The method of decellularization impacted gelation kinetics assessed by turbidimetric analysis. All hydrogels showed a fibrillary and porous structure determined by cryoSEM. Human corneal stromal cells were embedded in the hydrogels to assess cytotoxicity. SDS decellularization rendered cytotoxic hydrogels, while the other decellularization methods produced highly cytocompatible hydrogels. Freeze-thaw decellularization produced hydrogels with the overall best properties.
Similar content being viewed by others

Introduction
The extracellular matrix (ECM) is primarily composed of structural and regulatory proteins and polysaccharides and is generated and maintained by cells. Many cellular functions, such as proliferation, migration or differentiation are regulated by the ECM1. Each organ and tissue is composed of a distinctive ECM, in its biochemical composition and structural organization. The properties of ECM are important in the fields of tissue engineering and regenerative medicine, which often aim to replicate the composition and structure of the ECM. By using synthetic or natural materials, three-dimensional scaffolds can be fabricated to repair or restore damaged organs and tissues.
One popular approach to generating scaffolds that try to imitate the tissues or organs ECM characteristics is to use decellularization. This technique involves the removal of cellular components from a tissue so that only the ECM remains. Many methods have been examined for performing decellularization and these can be divided into three main categories: physical, chemical and biological2. Physical methods include freeze-thawing cycles3,4,5,6, high hydrostatic pressure7,8,9 or supercritical CO210,11,12. Chemical agents can involve ionic detergents, such as sodium dodecyl sulphate (SDS)13,14 or sodium deoxycholate15; non-ionic detergents, such as Triton X-10016; hypertonic or hypotonic salt solutions, such as sodium chloride17,18; and acids and bases, such as peracetic acid19 or ammonium hydroxide20. Enzymes such as trypsin, dispase and phospholipase A2 have been used as biological methods for decellularization21,22. Furthermore, nucleases, such as DNAse, are used to promote the fragmentation of residual DNA into <200 bp fragments in order to minimize immunological responses2. Extensive research has been completed to optimize these decellularization procedures to allow for maximal cell removal and minimal ECM damage for each tissue/organ.
One difficulty associated with some decellularized tissues is their limited potential for recellularization. For many tissue-engineering applications, healthy cells need to be embedded into the ECM to generate a functional and viable tissue. To overcome this problem decellularized organs and tissues can be transformed into hydrogels that allow cells to be encapsulated throughout their structure. These hydrogels can then be used as injectables for minimally invasive delivery into irregular spaces23,24,25,26,27,28 and for 3D printing of scaffolds29,30,31,32,33,34. Since the first report of ECM-derived hydrogels in 199835, over 70 papers have appeared in the literature describing the use of ECM-derived hydrogels from a wide variety of organs36. ECM-derived hydrogels have been under investigation to treat several medical conditions. These include type 1 diabetes, where the hydrogel provided a matrix to delivery cells to the pancreas27; myocardial infarction by replacing damaged cardiac tissue23, skin wounds37, and keratoconus by using the ECM to 3D bioprint a corneal stromal substitute38. Despite the increasing interest in such hydrogels, the effect of different decellularization methods on the final hydrogel characteristics has not been widely studied.
The aim of this study was to examine the impact of three different decellularization protocols on ECM-derived hydrogels obtained from porcine corneas. Two detergent-based techniques (SDS and Triton X-100) and a freeze-thaw cycling technique were used to decellularize corneas and hydrogels were fabricated from the resulting ECM. The impact of these decellularization protocols were evaluated in terms of biochemical composition, transparency, gelation kinetics, mechanical properties and cytocompatibility.
Results
Biochemical characterization of decellularized material
The biochemical composition of the fabricated ECM hydrogels was analysed. PicoGreen was used to quantify DNA remnants, collagen content was measured by hydroxyproline quantification and sulphated glycosaminoglycans (sGAG) were quantified by dimethylmethylene blue assay (DMMB). All decellularization methods led to a significant reduction in DNA when compared to the non-decellularized control, i.e. hydrogels from native corneas (Fig. 1A). Collagen levels remained constant in all treatments (Fig. 1B). sGAG levels were maintained when decellularization was performed with Triton or the freeze-thaw methods, while SDS resulted in significant loss of sGAG (Fig. 1C). Furthermore, histological examination appeared to validate these results (Fig. 1D). Staining with haematoxylin & eosin confirmed the absence of cell nuclei after decellularization. Dense collagen was observed after picro-sirius red staining across all samples. Alcian blue staining showed the presence of sGAG in all hydrogels with a noticeable reduction in staining for the SDS treated group.

Evaluation of decellularization of ECM-derived hydrogels: (A) Quantification of dsDNA, (B) Collagen, and (C) sGAG; *p < 0.05, **p < 0.01, ***p < 0.001; (D) histological examination of hydrogels, stained with haematoxylin and eosin, picro-sirius red and Alcian blue; black scale bar = 100 µm, white scale bar = 50 µm.
Further analysis of the composition of ECM-derived hydrogels was performed using SDS-PAGE and western blotting. SDS-PAGE showed the presence of collagen chains β, α1 and α2 for all conditions. Gamma chains were too heavy to be detected in a 7% polyacrylamide gel. Other lighter proteins were detected in the ECM-derived material lanes but not in a pure collagen type I control isolated from rat tail (Fig. 2). Immunodetection via western blot detected the presence of the corneal proteoglycan keratocan in all ECM-derived materials, independent of decellularization method, but not in rat tail derived collagen.

(A) Biochemical composition of ECM-derived hydrogels via SDS-PAGE (7%); (B) western blot against keratocan; M = molecular weight ladder.
Light transmittance
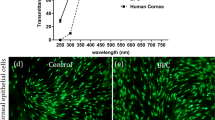
Since ECM from cornea was used to fabricate the hydrogels in this study, it is necessary to examine the transparency of the hydrogels since this is required for corneal tissue engineering. Transparency was measured by quantifying the light absorbed by the material and from this calculating the amount of light transmitted through each sample. As seen in Fig. 3, all hydrogels allowed light to pass through them, although hydrogels decellularized using SDS and the native hydrogels were cloudier in appearance. Quantitatively, all hydrogels presented at least 50% light transmittance at the end of the visible spectrum. Hydrogels decellularized using the freeze-thaw method were the most transparent with transmittance values above 70%. These values are only slightly lower than full thickness porcine corneas.

Transparency analysis of ECM-derived hydrogels: (A) Macroscopic appearance with hydrogels placed over printed text; (B) light transmittance quantification over the visible spectrum of light.
Gelation kinetics
Gelation kinetics of ECM hydrogels were analysed by turbidimetric analysis. This technique is based on the increased in turbidity, and thus absorbance, experienced during collagen self-assembly. All samples presented a sigmoidal profile and gelled after a lag period or tlag (Fig. 4). All treatments yielded hydrogels which started gelling after a longer lag phase than pure rat tail collagen (tlag 7.93 ± 0.55 minutes). Freeze-thawing produced the earliest gelling material of all treatments (tlag 16.43 ± 0.37 minutes), while SDS treated hydrogels took the longest to gel (tlag 27.53 ± 1.36 minutes). However, there was no statistically significant difference between the different speeds at which the ECM-derived materials gelled. All values are displayed in Table 1.

Gelation kinetics of ECM-derived hydrogels via turbidimetric analysis: (A) Raw values; (B) normalized data.
Rheology of ECM hydrogels
Rheology was utilized to assess mechanical characteristics of the hydrogels. Increasing shear rates were used to calculate the viscosity at 15 °C, quite below gelling temperature. Shear thinning properties were observed in all pre-gel solutions, regardless of decellularization treatment (Fig. 5A). Storage modulus (G′) and loss modulus (G′) were determined by following the gelation kinetics at 37 °C over time at a fixed frequency of 1 rad/s and 5% strain. All hydrogels had similar moduli values with no statistical significance among decellularization treatments (Fig. 5B). Only the Triton and the freeze-thaw groups were significantly weaker than the rat tail collagen hydrogels.

Rheology analysis of ECM-derived hydrogels: (A) Viscosity measurements at increasing shear rates; (B) storage modulus (G′) and loss modulus (G′); *p < 0.05.
Evaluation of hydrogel ultrastructure
CryoSEM was employed to study the structure of the hydrogels in the least disruptive way. Samples were snap frozen in nitrogen, sublimated, freeze-fractured and coated for SEM imaging. SEM confirmed the porous and fibrillar structure of the hydrogels, without evident differences between treatments (Fig. 6). Some areas displayed inhomogeneity in the density of fibres.

CryoSEM micrographs of ECM-derived hydrogels at ~1000x; scale bar = 10 µm.
Cytocompatibility
Human corneal stromal cells were embedded in the hydrogels to examine their cytocompatibility. Cell viability was assessed via calcein-acetoxymethyl ester and ethidium homodimer staining (Fig. 7A). After 1 day in culture, cells were highly viable in the Triton, freeze-thaw and native control hydrogel groups, while no viable cells were visible in the SDS treated group. Healthy cells presented an elongated morphology with small processes, indicating adhesion to the fibrillary architecture of the hydrogels. Over 5 days in culture, the hydrogels underwent significant contraction reflecting the ability of viable cells to actively attach and remodel the hydrogel (Fig. 7B,C). As expected from the viability assessment, the SDS hydrogel group did not undergo contraction. Hydrogels obtained from the SDS decellularization protocol were cytotoxic presumably due to inefficient washing after decellularization. To confirm this hypothesis the presence of detergent residues was determined using a methylene blue active substances (MBAS) assay, which is widely used in water quality control39. This assay is based on the binding of the cations in methylene blue with the anions from the detergent that are extracted into the organic phase when in contact with chloroform. Methylene blue extraction confirmed the presence of SDS remnants in the hydrogel thus would explain their cytotoxicity (Fig. 7D).

Cell activity in ECM-derived hydrogels: (A) Cell viability assessment (green = live, magenta = dead, scale bar = 200 µm); (B) macroscopic images of cell-laden hydrogels over time in culture; (C) quantification of hydrogel area over time; (D) quantification of methylene blue absorbance in the organic phase; *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
ECM-derived hydrogels offer great promise as biomaterials for tissue engineering as they can be delivered to the site in need in a minimally invasive manner, allow cell encapsulation prior to delivery, but can also allow for neighbouring cell recruitment. ECM hydrogel solutions have also been used as a bioink for 3D bioprinting several different tissues and organs29,40,41. In this study, ECM-derived hydrogels were obtained from corneas decellularized using three different methods. All decellularization methods decreased DNA significantly and retained collagen and other ECM components. All hydrogels were highly transparent, with the freeze-thaw group showing the best optical properties. Gelation kinetics were affected by the decellularization method employed but not the rheological properties. Hydrogels presented a porous and fibrillary structure. Hydrogels were highly cytocompatible when Triton and freeze-thawing methods were used for decellularization, but cytocompatibility was compromised when using SDS as decellularization agent. This work highlights the influence that the decellularization process has on final properties of ECM-derived hydrogels.
One of the main benefits of using ECM-derived hydrogels is the ability to retain multiple ECM components that may not be present in other natural or synthetic hydrogels and therefore more closely mimics the native tissues. Composition analysis of the digested materials via SDS-PAGE confirmed the presence of multiple ECM components when compared to rat tail collagen which just consists of collagen type I. Keratocan, a small leucine-rich proteoglycan almost exclusively found in the cornea, was detected in the ECM-derived hydrogels, irrespective of the decellularization method used. However, this study did show that the choice of decellularization technique is important with SDS decellularization retaining less sGAG than the other techniques tested. This removal of sGAG following decellularization has been previously reported for cornea14,18,42,43, cartilage29,44, ligament45 and adipose tissue29. While this study demonstrated that the ECM composition was affected by the decellularization technique used, other techniques may be used to identify more specific tissue or organ ECM components such as mass spectrometry46,47,48,49 or enzyme-linked immunosorbent assays (ELISA)32,33,50.
One potential limitation with using ECM-derived hydrogels is that GAGs and various collagen types, such as collagen type V have been shown to interfere in collagen type I self-assembly in vitro51,52,53. In vivo small leucine-rich proteoglycans, such as keratocan and decorin in the cornea, play an important role in collagen fibrillogenesis, in terms of collagen assembly nucleation and linear and lateral fibril growth54. Therefore, the difference in gelation kinetics between the commercially available collagen type I and the ECM-derived hydrogels can be explained by the presence of ECM components other than collagen type I. Studies from ECM-derived hydrogels from other sources have reported a delay in fibrillogenesis (lag phase) similar to what was shown here. For example, hydrogels obtained from demineralized and decellularized bone showed a short lag phase of around 9 minutes55, while myocardium ECM presented a long lag phase of 40 minutes56. Hydrogels obtained from urinary bladder matrix57, dermis19 and pancreas58 presented lag periods in a similar range to the ones reported in this study, between 15 and 25 minutes. Furthermore, the presence of detergent remnants might have an influence in the increased gelation time seen in SDS hydrogels. When we attempted to use concentrations above 0.1% SDS for decellularization, it was found that hydrogels could not be formed. This is in agreement with findings from Gaetani and colleagues who could not fabricate pancreas ECM-derived hydrogels when they used 1% SDS for decellularization58.
Pre-gel solutions presented shear thinning characteristics, i.e. viscosity decreases as shear rate increases. Values presented here are in accordance to those reported for ECM-derived hydrogels from myocardium56, dermis19, urinary bladder matrix57 skeletal muscle24 and cornea50. This characteristic offers the potential for these gels to be used as an injectable biomaterial and for their use as bioinks in 3D bioprinting29,30,31,32,33,34. Gelation profiles seen with turbidimetric analysis were also obtained when using rheology. Despite being more concentrated than the rat tail collagen hydrogels, the cornea ECM-derived hydrogels were softer. However, these values are in a similar range to the ones found in hydrogels derived from other tissues55,57. The values are lower than those reported for the storage and loss moduli of the native cornea, which are 2 kPa and 0.3 kPa, respectively59. Additional steps such as cross-linking60 may be required to increase the modulus of the hydrogels to match the native corneas.
In this study, cryoSEM was used to investigate the ultrastructure of the hydrogels. This technique is believed to be better at retaining the hydrogel’s structure compared to conventional SEM as the water present in the highly hydrated hydrogels is sublimated at extremely low temperatures61. The hydrogels obtained here were highly fibrillar and porous, which closely resembled the structure reported for ECM-derived hydrogels from other tissues, such as dermis19, myocardium56, demineralized bone55 and small intestinal submucosa35. These studies imaged the hydrogels using conventional SEM after glutaraldehyde fixation and critical point drying of the samples. Johnson and colleagues also described the presence of areas of higher fibre matrix density than others, which prevented implementation of automatized pore size quantification56.
In the current study, standard gelation parameters where used that can influence the hydrogels properties if modified. Johnson and colleagues studied the effect of temperature, ionic strength, pH and ECM concentration on the fibril architecture, mechanical properties and gelation kinetics of myocardium ECM-derived hydrogels56. They showed that no hydrogels could be formed at 4 °C and 22 °C, while at 37 °C they obtained robust hydrogels. Fibre diameter was not influenced by any of the conditions studied. Similar to our results, the authors reported areas of increased fibre density visualized by SEM. The effect that reduction of ionic strength to 0.5x PBS was striking as it increased mechanical properties and sped up gelation. pH did not influence any of the analysed parameters. Increase in ECM concentration increased mechanical properties and viscosity as reported for urinary bladder matrix57, bone55 and dermis19.
Furthermore, tissue origin plays an important role in hydrogel characteristics. It has been shown that porcine myocardium hydrogels retain more sGAG and have increased strength than healthy human myocardium hydrogels49. While using human tissues would ease the translation into the clinic as the issue of xenoimmunogenicity is avoided, sourcing healthy organs is difficult as these would be required for transplantation. However, for the cornea specifically, human corneas deemed unsuitable for transplantation due to low endothelial cell count, have the potential to be used to manufacture hydrogels. Decellularized porcine corneas have been used clinically as alternatives to donor grafts62,63, paving the path for other treatments based on ECM-derived materials.
Cells embedded within the hydrogels presented high viability and adopted a spindle morphology with multiple processes, indicating good adhesion to the fibres, except in the SDS group. Over the culturing period, the hydrogels contracted, as reported by Wolf and colleagues using dermal ECM-derived hydrogels19. This contraction effect was due to the traction forces exerted by the cells on the collagen fibrils. However, when cells died as in the SDS case, the hydrogels retained their shape and size. Depending on the application, the rate of contraction might limit the usefulness of these hydrogels without further cross-linking to strengthen them60 or using a cell culture condition that inhibits contractile behaviour64,65. Furthermore, contraction can also be inhibited when the hydrogel adheres to a material or is constrained at the edges66. When the hydrogel adheres to the tissue matrix, it will be less able to contract compared to a free-floating hydrogel in culture medium.
In this study, all steps were performed under sterile conditions so that no final sterilization method was required. However, for clinical purposes authorities may require terminal sterilization. It has been shown that common sterilization techniques such as electron beam, gamma irradiation or ethylene oxide inhibit gel formation when performed on the powder, but not when the lyophilized digest is treated12. Furthermore, sterilizing the lyophilized digest could increase the likelihood of translation into the clinic as a ready-to-use product, whereby the clinician can rehydrate the lyophilize digest with a basic salt balanced solution and let the hydrogel form in situ.
Future studies are required to assess the suitability of these hydrogels before they can be used for clinical applications. The hydrogels will need to be tested in vivo to evaluate the immunological response and ability to integrate with the surrounding tissue. The survival and functionality of the hydrogels would also need to be monitored. Should the hydrogel not induce any negative effects in vivo then the potential for hydrogels to replace damaged or diseased tissue could be explored using suitable animal models. For example in the cornea, the potential for ECM-derived hydrogels to replace tissue damaged due to trauma or diseases such as keratoconus could be explored. This is important due to a global shortage of donor corneas available for transplantation67.
In summary, here we demonstrated the importance that the decellularization method has on the final characteristics of ECM-derived hydrogels using corneas as tissue model. Similar phenomena would be expected for hydrogels derived from the ECM of other tissues although the precise decellularization protocol should be specific to the tissue type. We would therefore recommend that researchers or companies involved in the development of ECM-derived hydrogels examine different decellularization techniques to find the optimal approach for preparing their hydrogels.
Materials and Methods
Decellularization of porcine corneas
Porcine ocular globes were obtained from a local slaughterhouse. The remaining pieces of flesh were removed and, under aseptic conditions, the eyes were immersed in 2% iodine solution (Videne, Ecolab, Belgium) in sterile phosphate buffer saline (PBS) for one minute, gently rocking throughout. The eyes were subsequently washed twice in sterile PBS and the central corneal button was excised using scissors and cut into small pieces (2 mm × 2 mm, approximately). Three decellularization methods were tested:
- (a)
SDS (anionic detergent): each corneal button was immersed in 3 ml of 0.1% (w/v) sodium dodecyl sulphate (SDS, Sigma-Aldrich) solution for 72 hours under rotation. Solution was exchanged every 24 hours.
- (b)
Triton (non-ionic detergent): each corneal button was immersed in 3 ml of 1% (v/v) Triton X-100 (Sigma-Aldrich) solution for 72 hours under rotation. Solution was exchanged every 24 hours.
- (c)
Freeze-thaw (mechanical procedure): each corneal button was immersed in 5 ml sterile deionized H2O and placed in a −80 °C freezer for a minimum of 5 hours. Thereafter, they were let to thaw at room temperature. Once thawed, the solution was exchanged and the procedure repeated until 5 freeze-thaw cycles had been completed.
Afterwards, all corneas where subjected to a DNAse treatment for 1 hour at 37 °C under rotation. DNAse (Sigma-Aldrich) was used at a concentration of 10 U/ml prepared in 10 mM magnesium chloride buffer at pH 7.5. Corneas where extensively washed with sterile deionized water for 72 hours, with solution exchanged every 24 hours, under gentle rotation. Finally, decellularized corneas where dehydrated using a freeze drier and turned into powder by cryomilling (SPEX SamplePrep Freeze/Mill). Non-decellularized corneas were lyophilized and cryomilled to be used as controls (native).
Hydrogel formation
ECM hydrogels were prepared as previously described68. Briefly, ECM powder was dissolved in 1 mg/ml pepsin solution in 0.1 M hydrochloric acid at a concentration of 20 mg/ml and incubated for 72 hours at room temperature under slow rotation. Hydrogels were formed by neutralizing the solution with 1 N NaOH, balancing salt concentration using 10x PBS and incubating at 37 °C for one hour to induce fibrillation. Hydrogels had a final ECM concentration of 16 mg/ml. This process is depicted in Fig. 8. Rat tail collagen type 1 hydrogels were fabricated as described previously68 and used as a control for some studies.

Main steps in the fabrication of cornea ECM-derived hydrogels.
Biochemical quantification
Quantification of dsDNA, sGAG and collagen was performed after papain digestion of 100 µl hydrogels. DNA was quantified with the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen), following manufacturer’s specifications. sGAG quantification was performed using dimethylmethylene blue dye-binding assay (Blyscan, Biocolor). Collagen content was inferred by measuring the content of hydroxyproline using a chloramine T assay69,70.
Histology
Hydrogels were fixed in 4% paraformaldehyde and processed for wax embedding. 6 µm-thick slices of each sample were cut, attached to a glass slide, dewaxed, rehydrated and stained as follows. Haematoxylin & eosin was used to visualize any nuclei still present after decellularization. Slides were stained with Harris Haematoxylin (Sigma-Aldrich) for 4 minutes, followed by a 10-minute wash with running tap water. Then slides were immersed in acid alcohol for 30 seconds and washed with tap water for 5 minutes. Finally, slides were stained with Eosin Y (Sigma-Aldrich) for 2 minutes. Picro-sirius red was used to assess collagen distribution. Slides were stained with Sirius Red (Sigma-Aldrich) in a saturated aqueous solution of picric acid for one hour and then for one minute in 0.5% acetic acid. Alcian blue was used to assess sGAG content. Slides were stained with 1% Alcian Blue 8GX (Sigma-Aldrich) in 0.1 M HCl for 5 minutes followed by three 30-second washes in dH2O. After staining, all slides were dehydrated and coverslipped using DPX.
SDS-PAGE and western blot
7.5% polyacrylamide gels were cast and loaded with 50 µg of sample mixed with Laemmli buffer (1:1) that was previously boiled for 5 minutes. Gels were run for 90 minutes at 120 V. Gels were then stained with GelCode (Thermo-Fisher) following manufacturer’s directions. For western blot, 100 µg were loaded into precast 12% SDS-PAGE gels (Biorad) and run at 200 V for 45 minutes. Gels were transferred to a PVDF membrane using a semi-dry transfer system (Thermo-Fisher) with Pierce 1-step transfer buffer (Thermo-Fisher). The membrane was activated with methanol and then blocked overnight at 4 °C with 3% BSA. Primary antibody (keratocan, sc-66941, Santa Cruz) was used at a dilution of 1:200 in 3% BSA and incubated overnight at 4 °C. Three 5-minute washes with TBST were performed under agitation, and then the HRP-linked secondary antibody (A0545, Sigma-Aldrich) at a dilution of 1:1000 was incubated for an hour at room temperature. Another set of washing steps was carried out and then membranes were developed with Western Chemiluminescent HRP Substrate (Fisher Scientific). Membranes were imaged with GelDoc (Biorad).
Transparency
The macroscopic appearance and transparency of the gels was assessed by placing them over printed text. Subsequently, light transmittance was quantified. The absorbance of light at several wavelengths ranging from 350 to 700 nm was determined with a microplate reader (BioTek Synergy HTX). Deionized water was used as a baseline control. The transmittance of light was calculated using the following formula:
Gelation kinetics
Gelation kinetics was determined via turbidimetric spectrophotometric analysis, as described elsewhere57. Briefly, 100 µl hydrogels were casted into 96-well plates at 4 °C and inserted in a plate reader pre-heated at 37 °C (BioTek Synergy HTX). Absorbance at 405 nm wavelength was measured every 3 minutes for 90 minutes. Absorbance values were normalized with the following formula:
where NA is the normalized absorbance, A is the absorbance at any given time, A0 is the initial absorbance and Amax is the maximal absorbance.
The lag phase (tlag) was calculated by obtaining the linear portion of the curve and extrapolating the time value at which the normalized absorbance is 0. Similarly, t1/2 was determined as the time at which the normalized absorbance is 0.5. The slope of the linear portion of the curve determined the gelation speed (S).
Rheology
All rheological experiments were performed using a MCR 102 rheometer (Anton Paar, Austria) equipped with temperature controlling systems and using a 25 mm diameter parallel plate. Viscosity of pre-gel solutions was measured by performing a frequency sweep, from 0.1 to 1000 Hz, at 5% strain at 15 °C. Viscosity constants can be found in Supplementary Table S1. Storage and loss moduli were calculated with fixed frequency of 1 rad/s and 5% strain. Pre-gel solutions were applied at 4 °C and left equilibrate during 10 minutes, after which temperature was raised to 37 °C to induce gelation. Measurements where stopped once G’ values plateaued.
CryoSEM
Hydrogels were snap-frozen in nitrogen for 5 seconds, sublimated for 40 minutes at −100 °C and 10−5 Pa, freeze fractured and sputter coated with platinum for 20 seconds. These were then imaged with a scanning electron microscope at 5 kV (Ultra 2 Zeiss, Germany, with a Quorum Technologies CryoSEM Preparation System, UK).
Cell culture
Human corneal stromal cells were isolated and cultured as previously described in accordance with the Declaration of Helsinki64. The use of human cornea tissue with donor consent for isolating cells received ethical approval from the Trinity College Dublin, University of Dublin, School of Medicine Research Ethics Committee. 100,000 cells were embedded in each 100 µl hydrogel and cast in the wells of a 96-well plate. After gelation, hydrogels were released from the wells and transferred to 24-well plates. Constructs were fed every second day with low glucose DMEM (Hyclone) supplemented with 10% FBS, 100 U/ml Penicillin 100 µg/ml Streptomycin (both Gibco). Cell viability was assessed at day 1 and day 5 by staining the constructs with 2 µM calcein-acetoxymethyl ester and 4 µM ethidium homodimer-1 in PBS for 1 hour at 37 °C in a humidified incubator. Cells were then imaged via laser scanning confocal microscopy (Leica SP8). Furthermore, the shape of the hydrogels was monitored and images were taken daily over 5 days. The area of the hydrogels was calculated using Image J (NIH) and was plotted as the percentage of change of area with time.
Methylene blue active substances (MBAS) assay
MBAS assay was performed with some modifications from previously described methods71. A methylene blue (Sigma-Aldrich) solution was prepared in water to a final concentration of 250 µg/ml. 1 mg of cryomilled powder from each experimental group was mixed with 1 ml of distilled water and vortexed thoroughly for 1 minute and spun down for 30 seconds on a mini-centrifuge. 250 µl of this supernatant was mixed with 250 µl methylene blue solution and vortexed. Then 1 ml of chloroform was added, vortexed 3 times for 30 seconds and centrifuged for 1 minute using a benchtop centrifuge. A negative control was obtained using 250 µl distilled water and a positive control using 250 µl of 0.5% SDS solution. A phase separation was evident in all tests and visually the SDS group had a blue coloration in the organic (bottom) phase. This was further quantified by measuring the absorbance of the bottom phase at 665 nm using a plate reader (BioTek Synergy HTX).
Statistics analysis
GraphPad Prism Software 5.0 (GraphPad Software, Inc. La Jolla, CA, USA) was used to perform statistical analyses. All data are presented as the mean ± SD. One-way ANOVA with Tuckey post-hoc analyses were performed to determine statistical significance. Differences were considered to be statistically significant at p ≤ 0.05.
Change history
19 December 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Frantz, C., Stewart, K. M. & Weaver, V. M. The extracellular matrix at a glance. J. Cell Sci. 123, 4195–4200 (2010).
Crapo, P. M., Gilbert, T. W. & Badylak, S. F. An overview of tissue and whole organ decellularization processes. Biomaterials 32, 3233–3243 (2011).
Proulx, S. et al. Tissue engineering of feline corneal endothelium using a devitalized human cornea as carrier. Tissue Eng. Part A 15, 1709–18 (2009).
Ide, C., Tohyama, K., Yokota, R., Nitatori, T. & Onodera, S. Schwann cell basal lamina and nerve regeneration. Brain Res. 288, 61–75 (1983).
Utomo, L. et al. Preparation and characterization of a decellularized cartilage scaffold for ear cartilage reconstruction. Biomed. Mater. 10, 015010 (2015).
Rahman, S., Griffin, M., Naik, A., Szarko, M. & Butler, P. E. M. Optimising the decellularization of human elastic cartilage with trypsin for future use in ear reconstruction. Sci. Rep. 8, 3097 (2018).
Hashimoto, Y. et al. Ultrastructural analysis of the decellularized cornea after interlamellar keratoplasty and microkeratome-assisted anterior lamellar keratoplasty in a rabbit model. Sci. Rep. 6, 27734 (2016).
Santoso, E. G. et al. Application of detergents or high hydrostatic pressure as decellularization processes in uterine tissues and their subsequent effects on in vivo uterine regeneration in murine models. PLoS One 9, e103201 (2014).
Funamoto, S. et al. The use of high-hydrostatic pressure treatment to decellularize blood vessels. Biomaterials 31, 3590–3595 (2010).
Guler, S., Aslan, B., Hosseinian, P. & Aydin, H. M. Supercritical carbon dioxide-assisted decellularization of aorta and cornea. Tissue Eng. Part C Methods 23, 540–547 (2017).
Seo, Y., Jung, Y. & Kim, S. H. Decellularized heart ECM hydrogel using supercritical carbon dioxide for improved angiogenesis. Acta Biomater. 67, 270–281 (2018).
White, L. J. et al. The impact of sterilization upon extracellular matrix hydrogel structure and function. J. Immunol. Regen. Med. 2, 11–20 (2018).
Zilic, L. & Wilshaw, S.-P. & Haycock, J. W. Decellularisation and histological characterisation of porcine peripheral nerves. Biotechnol. Bioeng. 113, 2041–53 (2016).
Lynch, A. P., Wilson, S. L. & Ahearne, M. Dextran preserves native corneal structure during decellularization. Tissue Eng. Part C Methods 22, 561–572 (2016).
Price, A. P. et al. Automated decellularization of intact, human-sized lungs for tissue engineering. Tissue Eng. Part C Methods 21, 94–103 (2015).
Choi, J. S. et al. Bioengineering endothelialized neo-corneas using donor-derived corneal endothelial cells and decellularized corneal stroma. Biomaterials 31, 6738–6745 (2010).
Luo, H. et al. Construction of tissue-engineered cornea composed of amniotic epithelial cells and acellular porcine cornea for treating corneal alkali burn. Biomaterials 34, 6748–6759 (2013).
González-Andrades, M. et al. Generation of bioengineered corneas with decellularized xenografts and human keratocytes. Investig. Ophthalmol. Vis. Sci. 52, 215–220 (2011).
Wolf, M. T. et al. A hydrogel derived from decellularized dermal extracellular matrix. Biomaterials 33, 7028–7038 (2012).
Baptista, P. M. et al. The use of whole organ decellularization for the generation of a vascularized liver organoid. Hepatology 53, 604–617 (2011).
Chen, R.-N., Ho, H.-O., Tsai, Y.-T. & Sheu, M.-T. Process development of an acellular dermal matrix (ADM) for biomedical applications. Biomaterials 25, 2679–86 (2004).
Wu, Z. et al. The use of phospholipase A2 to prepare acellular porcine corneal stroma as a tissue engineering scaffold. Biomaterials 30, 3513–3522 (2009).
Singelyn, J. M. et al. Naturally derived myocardial matrix as an injectable scaffold for cardiac tissue engineering. Biomaterials 30, 5409–5416 (2009).
Ungerleider, J. L., Johnson, T. D., Rao, N. & Christman, K. L. Fabrication and characterization of injectable hydrogels derived from decellularized skeletal and cardiac muscle. Methods 84, 53–59 (2015).
Wang, J. Y. et al. Neurorestorative effect of urinary bladder matrix-mediated neural stem cell transplantation following traumatic brain injury in rats. CNS Neurol. Disord. Drug Targets 12, 413–425 (2013).
Zhang, L. et al. Effect of an inductive hydrogel composed of urinary bladder matrix upon functional recovery following traumatic brain injury. Tissue Eng. Part A 19, 1909–18 (2013).
Chaimov, D. et al. Innovative encapsulation platform based on pancreatic extracellular matrix achieve substantial insulin delivery. J. Control. Release 257, 91–101 (2017).
Lin, C.-Y., Liu, T.-Y., Chen, M.-H., Sun, J.-S. & Chen, M.-H. An injectable extracellular matrix for the reconstruction of epidural fat and the prevention of epidural fibrosis. Biomed. Mater. 11, 035010 (2016).
Pati, F. et al. Printing three-dimensional tissue analogues with decellularized extracellular matrix bioink. Nat. Commun. 5, 1–11 (2014).
Pati, F. et al. Biomimetic 3D tissue printing for soft tissue regeneration. Biomaterials 62, 164–175 (2015).
Jang, J. et al. Tailoring mechanical properties of decellularized extracellular matrix bioink by vitamin B2-induced photo-crosslinking. Acta Biomater. 33, 88–95 (2016).
Jang, J. et al. 3D printed complex tissue construct using stem cell-laden decellularized extracellular matrix bioinks for cardiac repair. Biomaterials 112, 264–274 (2017).
Skardal, A. et al. A hydrogel bioink toolkit for mimicking native tissue biochemical and mechanical properties in bioprinted tissue constructs. Acta Biomater. 25, 24–34 (2015).
Choi, Y. J. et al. 3D cell printing of functional skeletal muscle constructs using skeletal muscle-derived bioink. Adv. Healthc. Mater. 5, 2636–2645 (2016).
Voytik-Harbin, S. L., Brightman, A. O., Waisner, B. Z., Robinson, J. P. & Lamar, C. H. Small intestinal submucosa: a tissue-derived extracellular matrix that promotes tissue-specific growth and differentiation of cells in vitro. Tissue Eng. 4, 157–174 (1998).
Saldin, L. T., Cramer, M. C., Velankar, S. S., White, L. J. & Badylak, S. F. Extracellular matrix hydrogels from decellularized tissues: Structure and function. Acta Biomater. 49, 1–15 (2017).
Engel, H. et al. Investigation of Dermis-derived hydrogels for wound healing applications. Biomed. J. 38, 58–64 (2015).
Kim, H. et al. Shear-induced alignment of collagen fibrils using 3D cell printing for corneal stroma tissue engineering. Biofabrication 11, 035017 (2019).
George, A. L. & White, G. F. Optimization of the methylene blue assay for anionic surfactants added to estuarine and marine water. Environmental Toxicology and Chemistry 18, (1999).
Lee, H. et al. Development of liver decellularized extracellular matrix bioink for three-dimensional cell printing-based liver tissue engineering. Biomacromolecules 18, 1229–1237 (2017).
Kim, B. S., Kim, H., Gao, G., Jang, J. & Cho, D. W. Decellularized extracellular matrix: A step towards the next generation source for bioink manufacturing. Biofabrication 9, (2017).
Pang, K., Du, L. & Wu, X. A rabbit anterior cornea replacement derived from acellular porcine cornea matrix, epithelial cells and keratocytes. Biomaterials 31, 7257–7265 (2010).
Sasaki, S. et al. In vivo evaluation of a novel scaffold for artificial corneas prepared by using ultrahigh hydrostatic pressure to decellularize porcine corneas. Mol. Vis. 15, 2022–2028 (2009).
Elder, B. D., Eleswarapu, S. V. & Athanasiou, K. A. Extraction techniques for the decellularization of tissue engineered articular cartilage constructs. Biomaterials 30, 3749–3756 (2009).
Gratzer, P. F., Harrison, R. D. & Woods, T. Matrix alteration and not residual sodium dodecyl sulfate cytotoxicity affects the cellular repopulation of a decellularized matrix. Tissue Eng. 2975–2983, https://doi.org/10.1089/ten.2006.12.ft-234 (2006).
Odorico, J. S. et al. Extracellular matrix scaffold and hydrogel derived from decellularized and delipidized human pancreas. Sci. Rep. 8, 1–16 (2018).
DeQuach, J. A. et al. Simple and high yielding method for preparing tissue specific extracellular matrix coatings for cell culture. PLoS One 5, e13039 (2010).
Farnebo, S. et al. Design and characterization of an injectable tendon hydrogel: a novel scaffold for guided tissue regeneration in the musculoskeletal system. Tissue Eng. Part A 20, 1550–1561 (2014).
Johnson, T. D. et al. Human versus porcine tissue sourcing for an injectable myocardial matrix hydrogel. Biomater. Sci. 2, 735–744 (2014).
Kim, H. et al. Characterization of cornea-specific bioink: high transparency, improved in vivo safety. J. Tissue Eng. 10, 1–12 (2019).
Birk, D. E., Fitch, J. M., Babiarz, J. P., Doane, K. J. & Linsenmayer, T. F. Collagen fibrillogenesis in vitro: interaction of types I and V collagen regulates fibril diameter. J. Cell Sci. 95, (1990).
Pins, G. D., Christiansen, D. L., Patel, R. & Silver, F. H. Self-assembly of collagen fibers. Influence of fibrillar alignment and decorin on mechanical properties. Biophys. J. 73, 2164–2172 (1997).
Brightman, A. O. et al. Time-lapse confocal reflection microscopy of collagen fibrillogenesis and extracellular matrix assembly in vitro. Biopolymers 54, 222–234 (2000).
Zhang, G. et al. Genetic evidence for the coordinated regulation of collagen fibrillogenesis in the cornea by decorin and biglycan. J. Biol. Chem. 284, 8888–97 (2009).
Sawkins, M. J. et al. Hydrogels derived from demineralized and decellularized bone extracellular matrix. Acta Biomater. 9, 7865–7873 (2013).
Johnson, T. D., Lin, S. Y. & Christman, K. L. Tailoring material properties of a nanofibrous extracellular matrix derived hydrogel. Nanotechnology 22, (2011).
Freytes, D. O., Martin, J., Velankar, S. S., Lee, A. S. & Badylak, S. F. Preparation and rheological characterization of a gel form of the porcine urinary bladder matrix. Biomaterials 29, 1630–1637 (2008).
Gaetani, R. et al. Evaluation of different decellularization protocols on the generation of pancreas-derived hydrogels. Tissue Eng. Part C Methods 24, 697–708 (2018).
Hatami-Marbini, H. Viscoelastic shear properties of the corneal stroma. J. Biomech. 47, 723–728 (2014).
Ahearne, M. & Coyle, A. Application of UVA-riboflavin crosslinking to enhance the mechanical properties of extracellular matrix derived hydrogels. J. Mech. Behav. Biomed. Mater. 54, 259–267 (2016).
Schatten, H. Scanning electron microscopy for the life sciences. (Cambridge University Press, 2013).
Zhang, M.-C. M. et al. Lamellar keratoplasty treatment of fungal corneal ulcers with acellular porcine corneal stroma. Am. J. Transplant. 15, 1068–1075 (2015).
Zheng, J. et al. Short‐term results of acellular porcine corneal stroma keratoplasty for herpes simplex keratitis. Xenotransplantation e12509 (2019).
Fernández-Pérez, J. & Ahearne, M. Influence of biochemical cues in human corneal stromal cell phenotype. Curr. Eye Res. 44, 135–146 (2019).
Miotto, M. et al. 4D corneal tissue engineering: achieving time-dependent tissue self-curvature through localized control of cell actuators. Adv. Funct. Mater. 29, 1807334 (2019).
Ahearne, M. et al. Online monitoring of the mechanical behavior of collagen hydrogels: Influence of corneal fibroblasts on elastic modulus. Tissue Eng. - Part C Methods 16, (2010).
Gain, P. et al. Global survey of corneal transplantation and eye banking. JAMA Ophthalmol. 134, 167 (2016).
Ahearne, M. & Lynch, A. P. Early observation of extracellular matrix-derived hydrogels for corneal stroma regeneration. Tissue Eng. Part C Methods 21, 1059–1069 (2015).
Ignat’eva, N. Y. et al. Determination of hydroxyproline in tissues and the evaluation of the collagen content of the tissues. J. Anal. Chem. 62, 51–57 (2007).
Kafienah, W. & Sims, T. J. Biochemical methods for the analysis of tissue-engineered cartilage. Methods Mol. Biol. 238, 217–30 (2004).
Mathapati, S. et al. Qualitative and quantitative detection of sodium deoxycholic acid in decellularized tissue. Indian J. Thorac. Cardiovasc. Surg. 26, 129–131 (2010).
Acknowledgements
The research leading to these results has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 637460) and from Science Foundation Ireland (15/ERC/3269).
Author information
Authors and Affiliations
Contributions
J.F.P. designed and conducted experiments analysed data and wrote the manuscript. M.A. supervised the project and corrected the manuscript for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernández-Pérez, J., Ahearne, M. The impact of decellularization methods on extracellular matrix derived hydrogels. Sci Rep 9, 14933 (2019). https://doi.org/10.1038/s41598-019-49575-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49575-2
This article is cited by
-
Incorporation of GelMA/PEGDA into the Decellularized Cornea as a Potential Hybrid Scaffold for In Situ Repairing of Deep Anterior Corneal Defects
Journal of Polymers and the Environment (2024)
-
Immunogenicity of decellularized extracellular matrix scaffolds: a bottleneck in tissue engineering and regenerative medicine
Biomaterials Research (2023)
-
The effect of decellularization protocols on characterizations of thermoresponsive and light-curable corneal extracellular matrix hydrogels
Scientific Reports (2023)
-
Growth anisotropy of the extracellular matrix shapes a developing organ
Nature Communications (2023)
-
Biofabrication, biochemical profiling, and in vitro applications of salivary gland decellularized matrices via magnetic bioassembly platforms
Cell and Tissue Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
