
Abstract
Zirconium dioxide provides an exceptional prototype material for studying the redistribution of the polaron holes and its magnetic coupling with their nearby anions owning to the difference oxygen binding behavior in the monoclinic phase. Here, we perform a comprehensive study of the p-electron magnetism in the nitrogen doped 2 × 2 × 2 monoclinic ZrO2 based on spin-polarized density functional theory. Nitrogen substitutions make the system display half-metallic properties and the origin of room temperature ferromagnetism ascribes to the p-p coupling interaction between N 2p and the host 2p states. The charge density difference and Mülliken population analyses provide evidences of charge redistributions. Our results reveal that the polaron transfer may alter the magnetic properties and it is greatly facilitated ferromagnetic coupling if the polaron holes are localized around a single anion dopant.
Similar content being viewed by others

Introduction
The defect-induced magnetism involving only s- and p-electron elements have captured great interest and exhibited promising potential applications in spintronics devices. Doping 2p light elements (LE) into oxide semiconductors, what is known as a model system of diluted magnetic semiconductors (DMSs)1,2,3,4, the room temperature ferromagnetism (RTFM) can be achieved. For instance, RTFM was reported in C- or N-doped ZnO5,6,7,8, TiO29,10,11 thin films and Li-doped ZnO12 nanorods. Theoretically, possible ferromagnetism (FM) was also predicted in C-, or N-doped TiO213,14, ZnO15,16, CaO17,18 and MgO19,20,21,22 by density functional theory (DFT) or self-interaction correction (SIC) calculations. Compared to the conventional 3d transition-metal (TM)-doped DMSs23,24,25,26,27, LE-doped DMSs can provide a novel way to exclude the problem of clusters or secondary phases formed by doping 3d TM elements. Moreover, the spatially extended p states of the host and the impurities may able to extend the magnetic interaction to longer range. Various experimental and theoretical explorations have shown that the exchange interactions between the structural defects are believed to be responsible in establishing the magnetic moment. However, it remains obstacle that the relationship between valence band holes and origin of stable magnetic ordering in these materials. Therefore, a comprehensive understanding on the p-electron magnetism is essential for further exploring other potential DMSs.
The reasons we choose zirconium dioxide (ZrO2) as a prototypical DMS are as follows. (i) ZrO2 as a promising candidate for gate dielectric material28,29,30,31,32, has attracted much attention due to its relatively large dielectric constant (~25), wide band gap (5.1–7.8 eV), high breakdown field and good thermal stability. Introducing ferromagnetism into the nonmagnetic (NM) ZrO2 may open a new avenue for its applications. (ii) Due to difference oxygen binding behaviors in monoclinic ZrO2 (m-ZrO2), it is an exceptional prototype material for understanding of the relationship between electronic structure and the p-electron characters in these DMSs. Inspired by the prediction of Ostanin et al.33 that cubic ZrO2 should have a high Curie temperature (<500 K) by doping Mn, lots of efforts have focused on Mn-doped cubic ZrO2 but rare attentions were paid to LE-doped m-ZrO2, especially for doping N34,35,36.
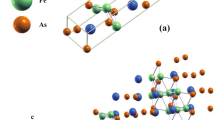
Unlike TiO2、 CeO2 and SnO2, oxygen sites in the m-ZrO2 can be divided into two types according to their binding behavior, one is bonded with three Zr atoms (labeled as O3) and the other is bonded with four Zr atoms (labeled as O4) [see Figs. 1(a) and 1(b)]. The projected density of states (PDOS) of the 2p orbitals for the O3 and O4 are presented in Fig. 1(d). At the first glance, the 2p orbitals of O3 can accommodate more holes than that in the 2p orbitals of O4 near the Fermi level. It implies that the O4 2p shell should be first fully occupied compare to the O3 2p shell while sufficient holes are injected. It can be anticipated that the redistribution of the induced holes and the interaction with their nearby atoms will have a significant impact on establishing the RTFM.

The geometries around (a) O3, (b) O4 and (c) Zr site in the m-ZrO2.
The cyan and red balls represent Zr and O atoms, respectively. (d) The PDOS for the O3 and O4 in the 2 × 2 × 2 ZrO2 supercell. The Fermi level is set to zero.
Results
Structure stability
Let us first consider a Zr32O63N supercell with a single N substituted at an O site, accompany a formation of a doped hole. After a structural relaxation, the N substituted at O3 site (NO3) model system is found to have a lower energy of 65.4 meV than that the N substituted at O4 site (NO4) model system. This should be due to the fact that NO3 has a more freedom to relax than the NO4. In addition, both spin-polarized and non-spin-polarized states are calculated to check the stability of the magnetization. The results reveal that both the model system of NO3 and that of NO4 favor the energy differences of about 387.5 and 293.1 meV between the spin-polarized state and the non-spin-polarized state respectively. The probability of incorporating a defect in a crystal is determined by the formation energy. The calculated formation energy of NO3 (or NO4) is 3.92 eV (or 3.99 eV) and −1.56 eV (or −1.49 eV) for the O-rich condition and Zr-rich condition, respectively.
Electronic properties
The calculated total density of states (TDOS) and PDOS for the N 2p, the neighboring Zr 4d and O 2p states are shown in Fig. 2. It is found there exist significant hybridization between the N 2p states and O 2p states around the Fermi level (EF), suggesting a strong p-p coupling interaction between them. Such a strong p-p interaction makes the energy levels near the EF spin split. The partially filled spin-down band split off from the fully occupied spin-up band via Hund exchange, resulting typical FM half-metallic character, which is important for the spintronics applications37. Additionally, nitrogen substituting for oxygen introduces some hole states peaked abound 1.0 eV above EF, which are mainly originated from N 2p, O 2p and Zr 4d orbitals. The total magnetic moment of the supercell is 1.00 μB corresponding to the single excess p electron provided by the N dopant. This is consistent with the previous results of N-doped ZnO15, TiO214, In2O338. The moments are mainly localized on the N 2p orbital (~0.86 μB), with minor contributions of the O (0.18 μB) and the neighboring Zr atoms is polarized antiferromagnetically with a magnetic moment of −0.04 μB (See the Supplementary Information for more details).

Calculated spin-resolved TDOS and PDOS of Zr32O63N with single N substituted at O3-type site.
The TDOS shows in (a). The (b), (c) and (d) plot the PDOS of N 2p states, the neighboring Zr 4d states and the neighboring O 2p states, respectively. The positive (negative) values represent the spin-up (spin-down) states. The vertical dotted line indicates the Fermi level at 0 eV.
The band structure calculations suggest that the localized holes induced by N dopant mediate the FM interaction. The carriers, i.e. holes, which were introduced by the substitution of N at O sites, reside not only on the N 2p orbitals but also on its near neighboring O atoms. The holes near each anion impurity tend to align parallel to the moment of the impurity ion under the p-d hybridization-like p-p coupling interaction5,39. Sufficient spin-polarized holes can mediate the spin alignment of the N dopant atoms, resulting in an indirect strong FM coupling among them. Therefore, the mechanism responsible for the FM coupling in the N-doped ZrO2 can be classified as the p-p coupling interaction between the impurity 2p states and the host 2p states.
Magnetic coupling
Next, we examine the magnetic coupling of these magnetic moments. To understand this issue, we calculate four nonequivalent structural configurations of Zr32O62N2, which are constructed by replacing two O atoms by N with various N-N distances. Here, the “dimer” of N-Zr-N configuration is only considered because we find the magnetic coupling to be negligible small when the N-N pair is well separated. The (i, j) configuration is adopted to denote an N-N pair, in which two O sites are replaced by N at i and j sites, as shown in Fig. 1(c). This amounts to the doping concentration of 3.13%. The magnetic coupling strength can be obtained from the energy difference between FM and AFM states (ΔEFM-AFM = EFM-EAFM). A positive value means the FM state is favored and vice versa. ΔE is the relative energy with respect to the (a, b) configuration which has the lowest total energy. The results are summarized in Table 1. It is found that the ΔE becomes smaller when the N-N distance gets larger and hence the doped N atoms do not have a tendency of clustering. It is noteworthy that the FM states always exhibit a lower energy than the AFM states except for the (a, d) configuration in which the N-N distance is 2.8 Å with the bond angle ∠N-Zr-N near 90°. It is well known that the Curie temperature in the mean field approximation (MFA) Tc is calculated as kBTc = (2/3c)ΔEFM-AFM, where kB is Boltzmann constant35,40,41. The MFA estimates Tc for DMSs with low concentrations within 5~10% errors. The Tc is always very lower than the room temperature, due to the short-range magnetic exchange interaction of deep impurity states in wide band-gap semiconductors, percolation of the ferromagnetic coupling is difficult to achieve for small concentrations. However, in the case of (a, b) configuration, the ΔEFM-AFM value reaches 372.7 meV, larger that of N-doped TiO214 and N-doped In2O338 which are known to be RTFM. Therefore, such a strong ferromagnetic interaction implies that the RTFM for N-doped ZrO2 is quite feasible considering the simple estimate. Though not shown here, our additional similar calculations for Zr32O62N2 with a long N-N distance (7.4 Å) reveal that the FM and AFM state are degenerate, whereas the total energy is 412.3 meV higher compared to the ground state of (a, b) configuration (See the Supplementary Information for more details). For comparison, let's focus on the largest and shortest N-N distance in the “dimer” of N-Zr-N configuration, corresponding to the (d, e) configuration and the ground state of (a, b) configuration, respectively.
For a deeply understanding of the physical origin of the ferromagnetism, the charge density difference of (a, b) and (d, e) configurations in FM states is calculated, as shown in Figs. 3(a) and 3(b), respectively. A weak covalent bonding can be seen between doped N atoms and its nearest-neighboring O atoms, suggesting that there exists a coupling interaction. Furthermore, in (d, e) configuration, the two doped N atoms which both N1 and N2 substituted at O3-type sites gain almost equivalent electrons from their neighboring Zr atoms. However, the situation is quite different in (a, b) configuration. The N4, where N substitutes O at O4-type site, 2p shell is almost completely filled as compared to the N3 where N substitutes O at O3-type site, just like the fully filled O2− 2p shell. These observations are consistent with the analysis in Fig. 1(d) that O4 2p shell is more easily to fully occupy than the O3 2p shell. This charge redistribution can also be quantitatively estimated by considering the Mülliken population analysis42. The charges of Zr, O and N atoms at different configurations are given in Table 2. To start with, we calculate the number of electrons on each atom in bulk ZrO2, which results in charge states of Zr and O atoms of +1.50e and −0.74e, respectively. Note that these values are far from the formal ones corresponding to the ideal ionic situation (Zr4+ and O2−), owning to the spatial division and the partial covalent feature of the Zr-O bonds. For (d, e) configuration, a Mülliken population analysis indicates that the local charges of N1 and N2 ions are −0.65e and −0.64e, whereas the neighboring of Zr and O remain essentially unchanged. Similar calculation for (a, b) configuration reveals that the N-N pair is strongly polarized, with charge states of −0.88e and −0.45e on the N4 and N3 sites, respectively. Given the above observations, the N1 (N2), N3 and N4 ions should have their electron configuration like N2− (s2p5), N1− (s2p4) and N3− (s2p6), respectively. Due to the coupling interaction between N 2p and O 2p, ppπ and ppσ states are formed. The schematic energy level diagram of the p-p hybridization between O2− and the above three kinds of N charge configurations (N2−, N1−, N3−) are illustrated in Figs. 3(c), 3(d) and 3(e). The nearly degenerate ppπ antibonding states are half-filled, 3/4 filled and fully filled, resulting 1.0 μB, 2.0 μB and 0.0 μB total magnetic moment, consistent with the DOS in Figs. 4(a)–4(c).

The charge density differences for the (d, e) and (a, b) configurations are shown in (a) and (b).
The schematic energy level diagram of pair interaction between O2− ions and N2−, N1− and N3− ions are illustrated in (c), (d) and (e), respectively.

The spin-resolved TDOS and PDOS for the (d, e) and (a, b) configurations.
(a) TDOS. The N 2p PDOS of (d, e) and (a, b) configurations in the FM states are illustrated in (b) and (c) respectively. The Fermi level is indicated by the dashed vertical line.
In order to further understand the nature of magnetic coupling in Zr32O62N2, the spin charge density (SCD) distribution of (a, b) and (d, e) configurations in the FM states and the AFM states are shown in Fig. 5. As can be seen, in (d, e) configuration, the p-like shape spin charge resides mainly on both the two N impurities and they have identical spin direction in the FM states while opposite direction in the AFM states. It indicates that there existed a strong FM coupling interaction induced by the N 2p orbitals [see also Fig. 4(b)]. Meanwhile, the lobes of N 2p orbitals are towards the lobes of O 2p orbitals to maximize the overlaps, which manifests that the p-p interaction does extend to the neighboring O ions, similar to the case of single N doping. In great contrast, for (a, b) configuration in the FM states, the main features are that the spin charge is mostly contributed by the N4 dopant and N-N pair coupling interaction nearly vanished [see also Fig. 4(c)]. This can be understood by noticing that the N-N distance is 4.4 Å, almost double of the distance in (d, e) configuration. Such a long distance effectively degenerate the N-N pair coupling interaction.

The isosurface SCD of (a, b) and (d, e) configurations in the FM states and the AFM states.
The cyan, red and blue balls represent Zr, O and N atoms, respectively. The green (yellow) isosurface correspond to positive (negative) spin charge densities. The isovalue is set to 0.02 e/Å3.
Discussion
Now we turn to address the question of what stabilizes the ferromagnetic ground state of (a, b) configuration. Spontaneous ferromagnetism can be evaluated according to the “Stoner Criterion”43,44: D(EF)J > 1, where D(EF) is the DOS at the Fermi level and J denotes the strength of the exchange interaction. The D(EF) is proportional to m3/2(EVBM-EF)1/2, where m is the effective mass and EVBM is the energy of the valence band maximum (VBM). Due to the stronger N3-Zr than N4-Zr bonds in the (a, b) configuration as indicated by their bond length (N3-Zr~2.05 Å, N4-Zr~2.22 Å), the N3 spin-up states are occupied and the occupation of the spin-down states will decrease. Thus the spin charge tend to accumulate around the N3 atom, which makes the defect level deeper and wave function more localized, resulting in a larger D(EF) and a larger spin-exchange splitting J of the band, as can be seen in Figure 4(a). The enhanced exchange splitting stabilizes the FM state, it can cause the system to be ground state. These results therefore indicate that it will be easier to obtain stable ferromagnetism if the injected holes are not shared by several neighboring anion atoms, but localize around a single anion. A similar situation was observed in the different system of C-doped ZnO45.
In summary, our ab initio calculation results indicate that the substitutional N induces spin-split impurity states near the Fermi level and leads to the half-metallic property. The total magnetic moment is found to be proportional to the number of injected holes in the supercell. The magnetic moments in this system are attributed to the strong p-p coupling interaction between the localized N 2p states and the neighboring O 2p states. An analysis from charge density difference and Mülliken population analysis was developed to explain the charge transfer due to the different binding behavior of N substituted at O sites. Our findings pave the way for interpreting further the nature of p-electron magnetism in LE-doped oxide DMSs.
Methods
ab initio calculations are performed by using density functional theory (DFT) with the plane-wave pseudopotential (PWP) approach as implemented in the Cambridge Sequential Total Energy Package (CASTEP)46. A generalized gradient approximation (GGA)47 in the form of Perdew-Burke-Ernzerhof (PBE) is used for the exchange correlation functional. The plane-wave cutoff energy is chosen to be 300 eV and a 2 × 2 × 2 Monkhorst-Pack k-point mesh is adopted for integrations over the Brillouin zone. The valence electron configurations for Zr, O and N are [Kr]4d25s2, [He]2s22p4 and [He]2s22p3, respectively. For structural relaxation, all the atomic positions are fully optimized until the atomic forces are smaller than 0.05 eV/Å. To study the low doping concentration of N-doped ZrO2, a 96-atom 2a × 2b × 2c supercell (Zr32O64) is used for all the calculations.
The defect formation energy Ef(nNO) of N-doped ZrO2 systems are calculated according to the following equation.

Here, Etot(ZrO2:N) is the total energy of the supercell with n oxygen atoms replaced by nitrogen atoms and Etot(ZrO2) is the total energy of ZrO2 perfect crystal in the same supercell. μO is the chemical potential of oxygen (μO ≤ μO2/2) and μN is the chemical potential of nitrogen in nitric oxide (NO)48.
For a supercell (ZrmOnNl) containing mZr atoms, nO and lN atoms, the charge density difference is obtained as follows:

where, ρ(ZrmOnNl) is the charge density of supercell ZrmOnNl, ρ(Zri), ρ(Oj) and ρ(Nk) are the charge density of isolated Zrm, On and Nk in the same supercell, respectively. According to the equation, the charge density difference gives a direct real-space visualization of the local charge redistribution due to the interactions of atoms.
To better understand the magnetic coupling of the Zr32O62N2 systems, SCD was used in all investigated systems and defined as:

where  and
and  are the spin up and spin down charge densities of the Zr32O62N2 system, respectively.
are the spin up and spin down charge densities of the Zr32O62N2 system, respectively.
References
Ohno, H. Making nonmagnetic semiconductors ferromagnetic. Science 281, 951 (1998).
Dietl, T., Ohno, H., Matsukura, F., Cibert, J. & Ferrand, D. Zener model description of ferromagnetism in zinc-blende magnetic semiconductors. Science 287, 1019 (2000).
Dietl, T., Ohno, H. & Matsukura, F. Hole-mediated ferromagnetism in tetrahedrally coordinated semiconductors. Phys. Rev. B 63, 195205 (2001).
Coey, J. M. D., Venkatesan, M., Stamenov, P., Fitzgerald, C. B. & Dorneles, L. S. Magnetism in hafnium dioxide. Phys. Rev. B 72, 024450 (2005).
Pan, H. et al. Room-Temperature Ferromagnetism in Carbon-Doped ZnO. Phys. Rev. Lett. 99, 127201 (2007).
Zhou, S. et al. Room temperature ferromagnetism in carbon-implanted ZnO. Appl. Phys. Lett. 93, 232507 (2008).
Yi, J. B. et al. Enhancement of room temperature ferromagnetism in C-doped ZnO films by nitrogen codoping. J. Appl. Phys. 105, 07c513 (2009).
Jinda, K., Tomar, M., Katiyar, R. S. & Gupta, V. Structural and magnetic properties of N doped ZnO thin films. J. Appl. Phys. 111, 102805 (2012).
Ye, X. J. et al. The magnetic property of carbon-doped TiO2 . Phys. Lett. A 373, 3684 (2009).
Gomez-Polo, C., Larumbe, S. & Pastor, J. M. Room temperature ferromagnetism in non-magnetic doped TiO2 nanoparticles. J. Appl. Phys. 113, 17b511 (2013).
Drera, G. et al. Enhancement of room temperature ferromagnetism in N-doped TiO2-x rutile: Correlation with the local electronic properties. Appl. Phys. Lett. 97, 012506 (2010).
Chawla, S., Jayanthi, K. & Kotnala, R. K. Room-temperature ferromagnetism in Li-doped p-type luminescent ZnO nanorods. Phys. Rev. B 79, 125204 (2009).
Yang, K., Dai, Y., Huang, B. & Whangbo, M.-H. On the possibility of ferromagnetism in carbon-doped anatase TiO2 . Appl. Phys. Lett. 93, 132507 (2008).
Tao, J. G. et al. Density functional study on ferromagnetism in nitrogen-doped anatase TiO2 . Appl. Phys. Lett. 95, 062505 (2009).
Shen, L. et al. Mechanism of ferromagnetism in nitrogen-doped ZnO: First-principle calculations. Phys. Rev. B 78, 073306 (2008).
Fang, D. Q., Zhang, R. Q., Zhang, Y. & Zhang, S. L. Effect of oxygen and zinc vacancies in ferromagnetic C-doped ZnO: Density-functional calculations. J. Magn. Magn. Mater. 354, 257 (2014).
Kenmochi, K., Seike, M., Sato, K., Yanase, A. & Katayama-Yoshida, H. New class of diluted ferromagnetic semiconductors based on CaO without transition metal elements. Jpn. J. Appl. Phys. 43, L934 (2004).
An Dinh, V., Toyoda, M., Sato, K. & Katayama-Yoshida, H. Exchange Interaction and Tc in Alkaline-Earth-Metal-Oxide-Based DMS without Magnetic Impurities: First Principle Pseudo-SIC and Monte Carlo Calculation. J. Phys. Soc. Jpn. 75, 093705 (2006).
Mavropoulos, P., Ležaić, M. & Blügel, S. Ferromagnetism in nitrogen-doped MgO: Density-functional calculations. Phys. Rev. B 80, 184403 (2009).
Slipukhina, I., Mavropoulos, P., Blügel, S. & Ležaić, M. Ferromagnetic Spin Coupling of 2p Impurities in Band Insulators Stabilized by an Intersite Coulomb Interaction: Nitrogen-Doped MgO. Phys. Rev. Lett. 107, 137203 (2011).
Pesci, M., Gallino, F., Di Valentin, C. & Pacchioni, G. Nature of Defect States in Nitrogen-Doped MgO. J. Phys. Chem. C 114, 1350 (2010).
Seike, M., Dinh, V. A., Fukushima, T., Sato, K. & Katayama-Yoshida, H. Self-Organized Nanostructures and High Blocking Temperatures in MgO-Based d(0) Ferromagnets. Jpn. J. Appl. Phys. 51, 050201 (2012).
Matsumoto, Y. et al. Room-temperature ferromagnetism in transparent transition metal-doped titanium dioxide. Science 291, 854 (2001).
Ohno, H. et al. (Ga,Mn)As: A new diluted magnetic semiconductor based on GaAs. Appl. Phys. Lett. 69, 363 (1996).
Sharma, P. et al. Ferromagnetism above room temperature in bulk and transparent thin films of Mn-doped ZnO. Nat. Mater. 2, 673 (2003).
Akai, H. Ferromagnetism and its stability in the diluted magnetic semiconductor (In,Mn)As. Phys. Rev. Lett. 81, 3002 (1998).
Reed, M. L. et al. Room temperature ferromagnetic properties of (Ga, Mn)N. Appl. Phys. Lett. 79, 3473 (2001).
Qi, W.-J. et al. Electrical and reliability characteristics of ZrO2 deposited directly on Si for gate dielectric application. Appl. Phys. Lett. 77, 3269 (2000).
Perkins, C. M. et al. Electrical and materials properties of ZrO2 gate dielectrics grown by atomic layer chemical vapor deposition. Appl. Phys. Lett. 78, 2357 (2001).
Fiorentini, V. & Gulleri, G. Theoretical evaluation of zirconia and hafnia as gate oxides for Si microelectronics. Phys. Rev. Lett. 89, 266101 (2002).
Ngai, T. et al. Electrical properties of ZrO2 gate dielectric on SiGe. Appl. Phys. Lett. 76, 502 (2000).
Mavrou, G. et al. Very high-kappa ZrO2 with La2O3 (LaGeOx) passivating interfacial layers on germanium substrates. Appl. Phys. Lett. 93, 212904 (2008).
Ostanin, S. et al. Mn-Stabilized Zirconia: From Imitation Diamonds to a New Potential High-TC Ferromagnetic Spintronics Material. Phys. Rev. Lett. 98, 016101 (2007).
Clavel, G., Willinger, M.-G., Zitoun, D. & Pinna, N. Manganese-doped zirconia nanocrystals. Eur. J. Inorg. Chem. 2008, 863 (2008).
Máca, F., Kudrnovský, J., Drchal, V. & Bouzerar, G. Magnetism without magnetic impurities in ZrO2 oxide. Appl. Phys. Lett. 92, 212503 (2008).
Zippel, J. et al. Defect-induced ferromagnetism in undoped and Mn-doped zirconia thin films. Phys. Rev. B 82, 125209 (2010).
Wolf, S. A. et al. Spintronics: A Spin-Based Electronics Vision for the Future. Science 294, 1488 (2001).
Guan, L. X., Tao, J. G., Huan, C. H. A., Kuo, J. L. & Wang, L. First-principles study on ferromagnetism in nitrogen-doped In2O3 . Appl. Phys. Lett. 95, 012509 (2009).
Sato, K., Dederichs, P. H., Katayama-Yoshida, H. & Kudrnovsky, L. Exchange interactions in diluted magnetic semiconductors. J. Phys.: Condens. Matter 16, S5491 (2004).
Kudrnovský, J. et al. Exchange interactions in III-V and group-IV diluted magnetic semiconductors. Phys. Rev. B 69, 115208 (2004).
Yang, K., Dai, Y. & Huang, B. First-principles characterization of ferromagnetism in N-doped SrTiO3 and BaTiO3 . Appl. Phys. Lett. 100, 062409 (2012).
Mulliken, R. S. Electronic Population Analysis on LCAO-MO Molecular Wave Functions. I. J. Chem. Phys. 23, 1833 (1955).
Stoner, E. C. Collective electron ferromagnetism. Proc. R. Soc. A 165, 0372 (1938).
Stoner, E. C. Collective electron ferromagnetism II Energy and specific heat. Proc. R. Soc. A 169, 339 (1939).
Peng, H. et al. Origin and Enhancement of Hole-Induced Ferromagnetism in First-Row d0 Semiconductors. Phys. Rev. Lett. 102, 017201 (2009).
Segall, M. D. et al. First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys.: Condens. Matter 14, 2717 (2002).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865 (1996).
Yan, Y., Zhang, S. B. & Pantelides, S. T. Control of Doping by Impurity Chemical Potentials: Predictions for p-Type ZnO. Phys. Rev. Lett. 86, 5723 (2001).
Acknowledgements
J. Li acknowledges the financial supports by the foundations under Grant Nos. 60578047, 2009CB929201, 2012CB934303, 13ZR1402600, 06DJ14007, 2011ZX02402, 11DZ2282200 and 2011ZRFL019. We thank Prof. S. Y. Wang for useful discussion and Prof. M. Xu & Prof. L. Y. Chen for effective backup.
Author information
Authors and Affiliations
Contributions
H.Z. carried out calculations, J.L. and F.G. took analyses and discussion. H.Z. and J.L. wrote the manuscript. K.C., X.Y. and S.C. discussed and reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhu, H., Li, J., Chen, K. et al. Nature of charge transport and p-electron ferromagnetism in nitrogen-doped ZrO2: An ab initio perspective. Sci Rep 5, 8586 (2015). https://doi.org/10.1038/srep08586
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08586
This article is cited by
-
Effect of Mg-doping in tailoring d0 ferromagnetism of rutile TiO2 compounds for spintronics application
Journal of Materials Science: Materials in Electronics (2021)
-
Magnetic properties of nitrogen-doped ZrO2: Theoretical evidence of absence of room temperature ferromagnetism
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
