
Abstract
Organic solar cells (OSCs) offer the possibility of harnessing the sun's ubiquitous energy in a low-cost, environmentally friendly and renewable manner. OSCs based on small molecule semiconductors (SMOSCs) – have made a substantial improvement in recent years and are now achieving power conversion efficiencies (PCEs) that match those achieved for polymer:fullerene OSCs. To date, all efficient SMOSCs have relied on the same fullerene acceptor, PCBM, in order to achieve high performance. The use of PCBM however, is unfavourable due to its low lying LUMO level, which limits the open-circuit voltage (VOC). Alternative fullerene derivatives with higher lying LUMOs are thus required to improve the VOC. The challenge, however, is to prevent the typical concomitant decrease in the short circuit current density (JSC) when using a higher LUMO fullerene. In this communication, we address the issue by applying methano indene fullerene, MIF, a bis-functionalised C60 fullerene that has a LUMO level 140 mV higher than PCBM, in solution processed SMOSCs with a well known small molecule donor, DPP(TBFu)2. MIF-based devices show an improved VOC of 140 mV over PC61BM and only a small decrease in the JSC, with the PCE increasing to 5.1% (vs. 4.5% for PC61BM).
Similar content being viewed by others

Introduction
Organic solar cells (OSCs) are becoming an increasingly viable low-cost, environmentally friendly and efficient means of generating electricity, with power conversion efficiencies exceeding 9% for single junction devices and 10% for tandem devices1,2. Within the past five years, small molecule OSCs (SMOSCs), where both the donor and acceptor are low molecular weight organic molecules, have made great strides with efficiencies now on par with their polymer:fullerene counterparts3,4. The improved performances obtained for SMOSCs has mainly come from the design of improved donor molecules that give higher absorption coefficients, better matching of the solar spectrum and enhanced miscibility with the fullerene acceptor, the latter previously being a strong limiting factor5,6,7. However, in all efficient solution processed BHJ SMOSCs to date the fullerene acceptor has been PCBM. In the case of polymer:fullerene OSCs a few examples of alternative fullerene derivatives do exist that allow higher efficiencies to be achieved by improving the open-circuit voltage (VOC)8,9. This improvement in VOC stems from the fullerene derivatives possessing a higher lying LUMO level, as the VOC is roughly proportional to the difference between the HOMO of the donor and LUMO of the acceptor10. Non-geminate recombination and the occupancy of the donor and acceptor density of states (DOS) will also have an effect on the VOC11,12,13,14, as can device architecture and the choice of selective contacts15,16.
Raising the LUMO level of a fullerene molecule can be achieved either through the addition of an electron donating moiety or by reducing the π-conjugation from 60-π electrons for C60 to 58-π electrons by the addition of a single adduct and to 56-π electrons by adding an additional adduct and so on17,18,19. Indene-C60 bisadduct (ICBA) is a good example of a bisadduct 56-π fullerene that improves the efficiency of P3HT based polymer:fullerene solar cells to 6.5%, which is significantly higher than the that of P3HT:PCBM, 3.8%, due to a 0.26 V increase in VOC8. However, except for P3HT and a few other examples20, most polymers blended with alternative fullerenes show significant losses in the short circuit current (JSC), which reduces or indeed negates the improvements in the overall power conversion efficiency (PCE, η) made through the increase in VOC21,22,23. Several explanations for this decrease in photocurrent have been presented. For example, Faist et al. found that a range of polymer donors blended with multiadduct fullerenes show low JSC values due either to reduced charge transfer efficiencies caused by small HOMO-HOMO or LUMO-LUMO offset energies between the donor and acceptor, or to poor charge collection efficiencies21. In the case of donor-acceptor combinations that produce VOC values that exceed 1 V, Hoke and co-workers found that the HOMO energy offsets between the donor and fullerene can prevent efficient hole transfer, which limits JSC22.
In SMOSC research, very few examples of non-PCBM acceptors exist. We previously found that, in the case of p-i-n devices based on a benzoporphyrin donor, higher efficiencies can be obtained using 58-π silylmethyl fullerene (SIMEF) acceptors rather than PC61BM, due to SIMEF having a higher lying LUMO level and thus generating a higher VOC, as well as an improved JSC due to the better packing of SIMEF24,25. A recent study by Palomares and co-workers attempted to understand the effect of replacing PC71BM with C70-based diphenylmethano fullerenes (DPMs) where the alkyl chains on the phenyl groups varied between 4 and 12 carbons26. In this study the donor molecule was a diketopyrolopyrole derivative, DPP(TBFu)2, which is a highly absorbing molecule (in solution it has a molar absorptivity of 64 000 M-1 cm-1 at 630 nm) that was developed by Nguyen and co-workers27. The effect of changing the fullerene adduct as well as the length of the terminal alkyl chains on the DPM adduct was found to affect the growth and crystallization of DPP(TBFu)2 domains, which led to significantly lower JSC values than the reference PC71BM devices. Although both DPM and PCBM were found to have the same LUMO level, VOC was higher for devices employing DPM fullerenes due to slower non-geminate recombination dynamics.
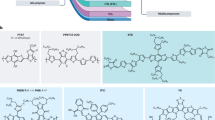
Here we apply a 56-π bis-functionalised fullerene (MIF), where one adduct is an indene group and the other is methanediyl group9, in SMOSCs using the well known DPP(TBFu)2 as a donor27, with the aim of improving the VOC due to the higher LUMO of this 56-π fullerene (−3.66 eV vs. 3.80 eV for PC61BM)9. The indene group provides for a good “alkyl chain-free” solubilizing group while the methanediyl adduct is the smallest possible adduct a fullerene can be functionalized with. This combination of adducts was chosen to reduce the conjugation of the fullerene in a manner that provides adequate solubility without using bulky adducts or adducts containing long alkyl chain substituents that could prevent, or at least limit, the formation of well-ordered DPP(TBFu)2 domains. Fig. 1 shows the device architecture and molecular structures and energy levels of DPP(TBFu)2 and MIF. We found that MIF does indeed improve VOC and maintains high JSC values, the latter being explained by the device morphology and DPP(TBFu)2 crystallinity, which are very similar to DPP(TBFu)2:PC61BM, as evidenced by atomic force microscopy (AFM) and X-ray diffraction (XRD).

Device architecture and molecular structure of DPP and MIF together with their HOMO-LUMO levels.
Results
Devices were prepared in a similar manner to previous studies with the following architecture: ITO/PEDOT:PSS/DPP(TBFu)2:Fullerene/LiF/Al26,28. A detailed description of device fabrication is provided in the methods section. The active layers were prepared from a 20 mg/ml solution with DPP(TBFu)2:fullerene ratios between 2:3 and 3:2. As in previous reports CHCl3 was the solvent of choice for DPP(TBFu)2:PC61BM27,28. However, a mixed solvent of CHCl3 and chlorobenzene (CB) was required for DPP(TBFu)2:MIF to sufficiently dissolve MIF, as it has low solubility in CHCl3 (1.5 wt%, see ESI for information on the solubility of MIF in various organic solvents). The ratio of CHCl3:CB was the same as that of DPP:MIF in each respective device, although we obtained similar device performances by adding as little as 10% CB to CHCl3 (ESI, Fig. S1). Solvent vapour annealing (SVA) was applied after spin-coating the active layer by introducing the substrates into a CH2Cl2 saturated container for 2 min. SVA is a proven method for producing highly crystalline donor domains for several small molecule donors and allows high fill factor (FF) values to be obtained26,28,29,30,31. Fig. 2 shows the UV-vis absorption spectra of DPP(TBFu)2:MIF films before and after the SVA step. After annealing a characteristic blue shift in the absorption spectrum was observed and is attributed to the aggregation and growth of DPP crystallites within the active layer28.

Absorption spectra of non-annealed (black) and annealed (blue) DPP(TBFu)2:MIF thin films.
J-V characteristics of DPP(TBFu)2:MIF with ratios of 2:3, 1:1 and 3:2 recorded under 1 sun simulated illumination (100 mW/cm2, AM 1.5G) and in the dark are shown in Fig. 3a and the figures of merit are presented in Table 1. A donor-acceptor ratio of 3:2 for DPP(TBFu)2:MIF was found to be optimum, providing for the higher JSC and improved series and shunt resistances (RS and RP, respectively), which led to improved FF values; the same ratio was also found to be optimum for both DPP(TBFu)2:PCBM devices in agreement with previous studies (see Table 1)27,28. All devices showed a VOC exceeding 1 V, the highest VOC being obtained using the 3:2 D:A ratio (1.03 V). Fig. 3b shows J-V curves of the best DPP:MIF device alongside reference DPP(TBFu)2:PC61BM and DPP(TBFu)2:PC71BM devices fabricated under the same conditions and D:A ratio to allow for the direct comparison between an efficient 58-π fullerene and for comparing with previous literature reports that utilised PC71BM27,28, respectively. We observe that the VOC for the 3:2 device is 140 mV higher than the DPP(TBFu)2:PC61BM and shows only a small decrease in JSC (9.52 mA/cm2 vs. 10.05 mA/cm2), while the FF is higher for the MIF based device (0.52 vs 0.50). DPP(TBFu)2:PC71BM have a similar VOC and FF to devices with the C60 analogue but have a higher JSC, as expected due to the better absorption characteristics of C70. In terms of efficiency, DPP(TBFu)2:MIF devices have a higher efficiency (5.1%) than both DPP(TBFu)2:PC61BM (4.5%) and DPP(TBFu)2:PC71BM (4.7%), due to the improved VOC, which arises from the increased energy difference between the LUMO of MIF and the HOMO of DPP(TBFu)2. MIF devices with a 3:2 ratio also have slightly higher FF values, in part due to the increased RP. However, the shape of the J-V curve can also be affected by geminate and non-geminate recombination32. Table 1 provides the figures of merit for all devices presented. Importantly, for this study and in the broader context, the optimum 3:2 DPP(TBFu)2:MIF device only shows a slight decrease in JSC with respect to DPP(TBFu)2:PC61BM devices while having a VOC over 1 V. The space charge limited current derived hole mobility of 3:2 DPP(TBF)2:MIF hole-only devices, 3.1 × 10−5 cm2 V−1 s−1, compared well with the equivalent PC61BM devices, 2.5 × 10−5 cm2 V−1 s−1, which is consistent with the high JSC values obtained for the MIF devices.

J-V characteristics measured under standard 1 sun conditions (AM 1.5 G, 100 mW/cm2) (solid lines) and dark conditions (dashed lines) for (a) DPP(TBFu)2:MIF devices with the corresponding DPP(TBFu)2:MIF ratios: 2:3 (orange, squares), 1:1 (blue, triangles) and 3:2 (red, circles) and (b) 3:2 DPP(TBFu)2:fullerene devices, where the fullerene is MIF (red, circles), PC61BM (blue, squares) or PC71BM (black, triangles).
Controlling the morphology of BHJ films is key to obtaining high efficiency devices and has been well exemplified in previous DPP(TBFu)2 studies26,28,33,34,35. Poor compatibility between donor and acceptor molecules, which can be identified by imaging non-annealed films, will prevent the formation of a well-ordered BHJ that is necessary for efficient charge generation, separation and extraction26. We thus investigated the morphology and crystallinity of DPP(TBFu)2:MIF BHJ films and compared them to DPP(TBFu)2:PC61BM. AFM images of annealed and non-annealed DPP(TBFu)2:MIF films are shown in Fig. 4 that demonstrate the good film formation between DPP(TBFu)2 and MIF. Non-annealed films show excellent miscibility between both molecules (Fig. 4a) and produce quite flat films (rms roughness = 0.88 nm) with almost no sign of aggregation. Subjecting the active layer to 2 min of SVA in a saturated CH2Cl2 environment led to a roughening of the surface (rms roughness = 1.25 nm) due to crystallization of DPP(TBFu)2 (Fig. 4b), which was observed using out-of-plane X-ray diffraction on the active layers (vide infra). The corresponding images of DPP:PC61BM are shown in Fig. 4c and 4d. One significant difference to note between the morphology of both blends is that the as-cast DPP(TBFu)2:PC61BM film shows considerable aggregation compared to DPP(TBFu)2:MIF and thus a much rougher surface (rms roughness = 2.04 nm). SVA allows the molecules to re-orientate and the resulting morphology shows no signs of aggregation and the roughness increases slightly (rms roughness = 2.17 nm) due to the more crystalline nature of the films as opposed to aggregates.

AFM images (5 × 5 μm) of 3:2 DPP(TBFu)2:MIF films (a,b) and DPP(TBFu)2:PC61BM (c,d) before (a,c) and after (b,d) solvent vapour annealing.
The XRD diffractograms of the DPP:MIF and DPP:PC61BM films are shown in Fig. 5 and show a peak at 2θ = 6.19° and 6.09°, respectively, corresponding to an inter-plane spacing of 14.3 Å and 14.5 Å, respectively, which is characteristic of pure DPP(TBFu)2, as seen in previous studies27,28. The average crystallite size for DPP(TBFu)2 in the annealed DPP(TBFu)2:MIF film, calculated using the Scherrer equation, was 17.9 nm36, which is very similar to what we calculated for DPP(TBFu)2 blended with PC61BM, 17.8 nm. There was, however, a difference in the overall crystallite volume observed between DPP(TBFu)2:MIF and DPP(TBFu)2:PC61BM films, with the latter having a higher volume (Fig. 5). No peaks corresponding to MIF were observed. The similarities observed between the AFM and XRD data of DPP(TBFu)2:MIF and DPP(TBFu)2:PC61BM BHJ films help explain the high hole mobility and JSC observed in optimum DPP(TBFu)2:MIF devices, especially if we consider what was observed by Viterisi et al. for DPP(TBFu)2 blended with DPM fullerene derivatives, for example, where the morphology of the active layer, as well as the crystallinity of DPP(TBFu)2, was significantly affected by the DPM fullerene26.

Out of plane XRD diffractograms of SVA treated DPP(TBFu)2:MIF and DPP(TBFu)2:PC61BM thin films on ITO/PEDOT:PSS.
Discussion
Putting these results into the perspective of previous reports of polymer:fullerene OSCs, very few fullerenes, in particular fullerenes with higher lying LUMO levels, seem to work well with polymers having a lower band-gap than P3HT. The LUMO and HOMO offsets may indeed explain some of the poorer performances observed but the miscibility between donor and acceptor and crystalline properties (both individual and collective) are also key factors that define the device performance. Many efficient low-bandgap polymers for example form amorphous domains, which may prevent optimum charge transport, as already suggested by Faist et al.21 DPP(TBFu)2 has a lower band-gap than P3HT and lower lying frontier molecular orbitals yet still maintains high JSC when MIF is the acceptor, meaning that energetic offsets between HOMO-HOMO and LUMO-LUMO levels at the donor-acceptor interface must be sufficiently large so as not to affect charge separation. The domain size of DPP(TBFu)2 must also be close to optimum in these devices to allow a high number of excitons to reach the D-A interface and indeed seems to be, with the average crystallite size being almost identical to that of DPP(TBFu)2:PC61BM devices. The total crystalline volume of the donor is, however, lower in DPP(TBFu)2:MIF active layers, which may explain the slight decrease in JSC. Furthermore, the AFM topography images show good intermixing of DPP(TBFu)2 and MIF and is again similar to what we observe for DPP(TBFu)2:PC61BM. Moving on from this study, it will be interesting to expand the application of MIF with other low bandgap small molecule and polymeric donors in order to gain a deeper understanding of the key factors that define high performance donor:fullerene BHJ solar cells.
In conclusion, we have applied MIF, a 56-π fullerene containing indene and methanediyl adducts, with a small molecule donor (DPP(TBFu)2) for the first time and obtained a power conversion efficiency of 5.1%. Reference DPP(TBFu)2:PC61BM and DPP(TBFu)2:PC71BM devices show lower performances of 4.5% and 4.7%, respectively. The higher efficiency of DPP(TBFu)2:MIF arises from the higher VOC achieved, which stems from MIF's higher lying LUMO level. Furthermore, no considerable changes in JSC or FF are observed between MIF and the PCBM reference devices, which can be understood by the excellent miscibility between DPP and MIF and the ability for DPP(TBFu)2 to form well ordered crystalline domains during SVA of the BHJ, as observed by AFM and XRD. The results obtained in this study provide key insights into the importance of optimizing both the electronic and molecular structure of fullerene derivatives to allow higher VOC values to be obtained without impacting upon the morphology of the donor molecule, which can limit the JSC.
Methods
General notes
DPP(TBFu)2 was purchased from Lumtec and further purified by silica gel column chromatography using a toluene:hexane eluent (9:1). MIF was synthesized and purified following our previously reported method9. Briefly, under a nitrogen atmosphere, a solution of C60(CH2) (300 mg, 0.409 mmol) and indene (1.14 mL, 9.81 mmol) in 1,2-dichlorobenzene (60 mL) was refluxed for 20 h. After removal of solvent by vacuum distillation, the obtained solid was subjected to silica gel column chromatography (eluent, CS2/hexane = 1/2 to 1/0) to remove starting materials. Gel permeation chromatography was then carried out to isolate MIF.
Device Fabrication
Devices were fabricated with the architecture: ITO/PEDOT:PSS/DPP(TBFu)2:Fullerene/LiF/Al. First, patterned indium-doped tin oxide (ITO) substrates (155 nm, 9 Ω/□) were sonicated in acetone for 15 min followed by two additional 15 min sonication cycles in isopropanol. Next, the substrates were dried under a stream of nitrogen and then subjected to 20 min UV/O3 treatment. PEDOT:PSS (Clevios AI4083) was spin-coated onto the clean ITO substrates at a rate of 3000 rpm for 30 sec. Annealing of PEDOT:PSS films was first done in air at 120°C and then in a N2 filled glovebox at 130°C for an additional 5 min. Active layers were then deposited by spin-coating at a rate of 3000 rpm for 60 sec. The donor:acceptor ratio (wt:wt) for DPP(TBFu)2:MIF devices varied between 2:3 and 3:2 for optimization purposes and had a total concentration of 20 mg/ml in CHCl3:chlorobenzene (CB) solution, where the ratio of each solvent (vol:vol) mirrored that of the active layer, i.e. 2:3 MIF:DPP(TBFu)2 was dissolved in a 2:3 CHCl3:CB solution. CB was required for dissolving MIF, however was not necessary for the PCBM reference devices. DPP(TBFu)2:PC61BM and DPP(TBFu)2:PC71BM devices had a ratio of 3:2 and the same concentration as the MIF-based devices (20 mg/ml) in CHCl3. Active layer thickness was approx. 90 nm as measured using a step-profiler. Solvent vapour annealing (SVA)14, which consisted of placing one substrate at a time in a sealed vessel saturated with CH2Cl2 for 2 min, was applied to each active layer after spin-coating. Following SVA the substrates were placed in an evaporator chamber where a 0.8 nm layer of LiF was first deposited followed by a 100 nm layer of Al. The pressure of the evaporation chamber never exceeded 5 × 10−5 mbar during deposition. Devices were sealed in a N2 rich environment using a UV curable epoxy before measuring their photovoltaic characteristics. Hole-only devices were fabricated in the same manner as normal devices except that a MoO3(8 nm)/Au (100 nm) top electrode was used.
Device Characterization
Current-voltage (J-V) characteristics were measured by software-controlled source meter (Keithley 2400) in dark conditions and 1 sun AM 1.5 G simulated sunlight irradiation (100 mW/cm2) using a solar simulator (EMS-35AAA, Ushio Spax Inc.), which was calibrated using a silicon diode (BS-520BK, Bunkokeiki).
Film Characterization
UV-visible absorption spectra were measured on JASCO V-670 spectrometer (Nihon bunko). Atomic force microscopy images were recorded using a Bruker Multimode atomic force microscope operating in tapping mode (Si probes, nominal frequency 70 kHz). Out-of-plane X-ray diffraction was carried out on a Rigaku Smartlab diffractometer using Cu-Kα radiation operating with a power of 9 kW (45 kV, 200 mA). The diffraction pattern of each sample was recorded between an angular 2θ of 2 and 14° at 0.5° increments, the duration of which were 3 sec.
References
He, Z. et al. Enhanced power-conversion efficiency in polymer solar cells using an inverted device structure. Nat. Photon. 6, 591–595 (2012).
You, J. et al. A polymer tandem solar cell with 10.6% power conversion efficiency. Nat. Commun. 4, 1446 (2013).
Kyaw, A. K. K. et al. Efficient solution-processed small-molecule solar cells with inverted structure. Adv. Mater. 25, 2397–2402 (2013).
Liu, Y. et al. Solution-processed small-molecule solar cells: breaking the 10% power conversion efficiency. Sci. Rep. 3, 3356 (2013).
Chen, Y., Wan, X. & Long, G. High performance photovoltaic applications using solution-processed small molecules. Acc. Chem. Res. 46, 2645–2655 (2013).
Roncali, J., Leriche, P. & Blanchard, P. Molecular materials for organic photovoltaics: small is beautiful. Adv. Mater. 26, 3821–3838 (2014).
Walker, B., Kim, C. & Nguyen, T.-Q. Small molecule solution-processed bulk heterojunction solar cells. Chem. Mater. 23, 470–482 (2011).
Zhao, G., He, Y. & Li, Y. 6.5% Efficiency of polymer solar cells based on poly(3-hexylthiophene) and indene-C60 bisadduct by device optimization. Adv. Mater. 22, 4355–4358 (2010).
Matsuo, Y. et al. Addition of dihydromethano group to fullerenes to improve the performance of bulk heterojunction organic solar cells. Adv. Mater. 25, 6266–6269 (2013).
Qi, B. & Wang, J. Open-circuit voltage in organic solar cells. J. Mater. Chem. 22, 24315–24325 (2012).
Bisquert, J. & Garcia-Belmonte, G. On voltage, photovoltage and photocurrent in bulk heterojunction organic solar cells. J. Phys. Chem. Lett. 2, 1950–1964 (2011).
Credgington, D. & Durrant, J. R. Insights from transient optoelectronic analyses on the open-circuit voltage of organic solar cells. J. Phys. Chem. Lett. 3, 1465–1478 (2012).
Garcia-Belmonte, G. et al. Influence of the intermediate density-of-states occupancy on open-circuit voltage of bulk heterojunction solar cells with different fullerene acceptors. J. Phys. Chem. Lett. 1, 2566–2571 (2010).
Sánchez-Díaz, A., Izquierdo, M., Filippone, S., Martin, N. & Palomares, E. The origin of the high voltage in DPM12/P3HT organic solar cells. Adv. Funct. Mater. 20, 2695–2700 (2010).
Ratcliff, E. L. et al. Investigating the influence of interfacial contact properties on open circuit voltages in organic photovoltaic performance: work function versus selectivity. Adv. Energy Mater. 3, 647–656 (2013).
Ryan, J. W., Kirchartz, T., Viterisi, A., Nelson, J. & Palomares, E. Understanding the effect of donor layer thickness and a MoO3 hole transport lyer on the open-circuit voltage in squaraine/C60 bilayer solar cells. J. Phys. Chem. C 117, 19866–19874 (2013).
Matsuo, Y. Design concept for high-LUMO-level fullerene electron-acceptors for organic solar cells. Chem. Lett. 41, 754–759 (2012).
Li, Y. Fullerene-bisadduct acceptors for polymer solar cells. Chem. Asian J. 8, 2316–2328 (2013).
Li, C.-Z., Yip, H.-L. & Jen, A. K. Y. Functional fullerenes for organic photovoltaics. J. Mater. Chem. 22, 4161–4177 (2012).
Huang, J.-H. et al. The investigation of donor-acceptor compatibility in bulk-heterojunction polymer systems. Appl. Phys. Lett. 103, 043304 (2013).
Faist, M. A. et al. Understanding the reduced efficiencies of organic solar cells employing fullerene multiadducts as acceptors. Adv. Energy Mater. 3, 744–752 (2013).
Hoke, E. T. et al. Recombination in polymer:fullerene solar cells with open-circuit voltages approaching and exceeding 1.0 V. Adv. Energy Mater. 3, 220–230 (2013).
Ahme, H., Lee, M., Im, C. & Würfel, U. Influence of the acceptor on electrical performance and charge carrier transport in bulk heterojunction solar cells with HXS-1. J. Phys. Chem. C 118, 3386–3392 (2014).
Matsuo, Y. et al. Columnar structure in bulk heterojunction in solution-processable three-layered p-i-n organic photovoltaic devices using tetrabenzoporphyrin precursor and silylmethyl[60]fullerene. J. Am. Chem. Soc. 131, 16048–16050 (2009).
Tanaka, H. et al. An amorphous mesophase generated by thermal annealing for high-performance organic photovoltaic devices. Adv. Mater. 24, 3521–3525 (2012).
Fernandez, D. et al. Small molecule BHJ solar cells based on DPP(TBFu)2 and diphenylmethanofullerenes (DPM): linking morphology, transport, recombination and crystallinity. Nanoscale 6, 5871–5878 (2014).
Walker, B. et al. Nanoscale phase separation and high photovoltaic efficiency in solution-processed, small-molecule bulk heterojunction solar cells. Adv. Funct. Mater. 19, 3063–3069 (2009).
Viterisi, A., Gispert-Guirado, F., Ryan, J. W. & Palomares, E. Formation of highly crystalline and texturized donor domains in DPP(TBFu)2:PC71BM SM-BHJ devices via solvent vapour annealing: implications for device function. J. Mater. Chem. 22, 15175–15182 (2012).
Sun, K. et al. The role of solvent vapor annealing in highly efficient air-processed small molecule solar cells. J. Mater. Chem. A 2, 9048–9054 (2014).
Wessendorf, C. D. et al. Efficiency improvement of solution-processed dithienopyrrole-based A-D-A oligothiophene bulk-heterojunction solar cells by solvent vapor annealing. Adv. Energy Mater. 4, 1400266 (2014).
Viterisi, A. et al. Unambiguous determination of molecular packing in crystalline donor domains of small molecule solution processed solar cell devices using routine X-ray diffraction techniques. J. Mater. Chem. A 2, 3536–3542 (2014).
Dibb, G. F. A., Jamieson, F. C., Maurano, A., Nelson, J. & Durrant, J. R. Limits on the fill Factor in organic photovoltaics: distinguishing nongeminate and geminate recombination mechanisms. J. Phys. Chem. Lett. 4, 803–808 (2013).
He, M., Wang, M., Lin, C. & Lin, Z. Optimization of molecular organization and nanoscale morphology for high performance low bandgap polymer solar cells. Nanoscale 6, 3984–3994 (2014).
Brabec, C. J., Heeney, M., McCulloch, I. & Nelson, J. Influence of blend microstructure on bulk heterojunction organic photovoltaic performance. Chem. Soc. Rev. 40, 1185–1199 (2011).
Fitzner, R. et al. Interrelation between crystal packing and small-molecule organic solar cell performance. Adv. Mater. 24, 675–680 (2012).
Scherrer, P. Nachr Ges Wiss Goettingen, Math-Phys Kl, 98–100 (1918).
Acknowledgements
This work was supported by the Funding Program for Next-Generation World-Leading Researchers. We thank Dr. Takafumi Nakagawa and Hiroshi Inada for synthesizing and/or purifying the active layer materials. Aurelien Viterisi is gratefully acknowledged for advice regarding DPP(TBFu)2 purification as well as for informative and interesting discussions.
Author information
Authors and Affiliations
Contributions
J.W.R. and Y.M. conceived the work. J.W.R. designed and performed all experiments and wrote the manuscript. Y.M. designed the concept to have high open-circuit voltage and modified the manuscript. Both authors discussed and agreed on the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ryan, J., Matsuo, Y. Increased Efficiency in Small Molecule Organic Solar Cells Through the Use of a 56-π Electron Acceptor – Methano Indene Fullerene. Sci Rep 5, 8319 (2015). https://doi.org/10.1038/srep08319
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08319
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
